

Critical roles for interleukin 1 and tumor necrosis factor alpha in antibody-induced arthritis
- PMID: 12093872
- PMCID: PMC2194010
- DOI: 10.1084/jem.20020439
Critical roles for interleukin 1 and tumor necrosis factor alpha in antibody-induced arthritis
Abstract
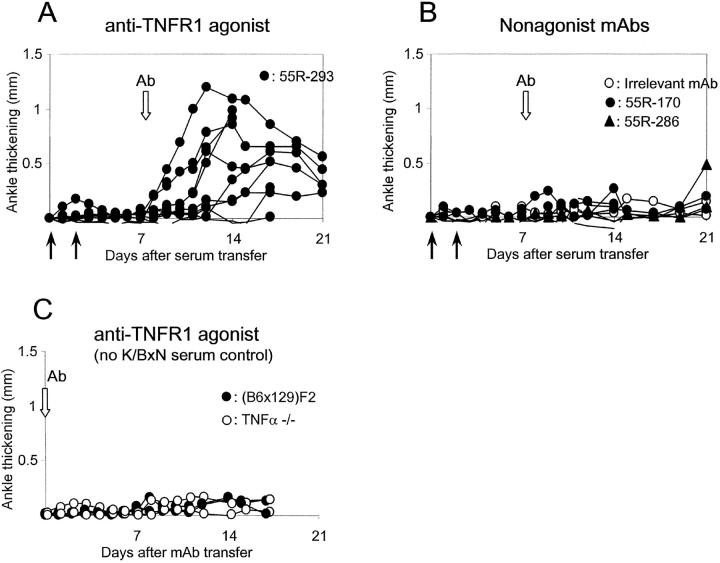
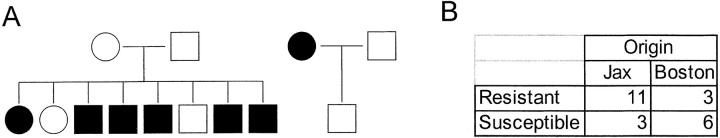
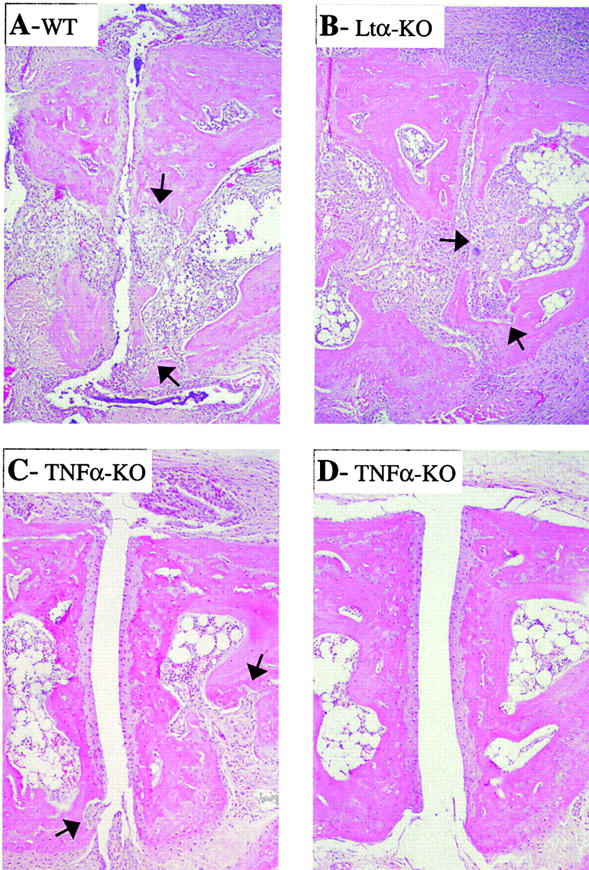
In spontaneous inflammatory arthritis of K/BxN T cell receptor transgenic mice, the effector phase of the disease is provoked by binding of immunoglobulins (Igs) to joint surfaces. Inflammatory cytokines are known to be involved in human inflammatory arthritis, in particular rheumatoid arthritis, although, overall, the pathogenetic mechanisms of the human affliction remain unclear. To explore the analogy between the K/BxN model and human patients, we assessed the role and relative importance of inflammatory cytokines in K/BxN joint inflammation by transferring arthritogenic serum into a panel of genetically deficient recipients. Interleukin (IL)-1 proved absolutely necessary. Tumor necrosis factor (TNF)-alpha was also required, although seemingly less critically than IL-1, because a proportion of TNF-alpha-deficient mice developed robust disease. There was no evidence for an important role for IL-6. Bone destruction and reconstruction were also examined. We found that all mice with strong inflammation exhibited the bone erosion and reconstruction phenomena typical of K/BxN arthritis, with no evidence of any particular requirement for TNFalpha for bone destruction. The variability in the requirement for TNF-alpha, reminiscent of that observed in treated rheumatoid arthritis patients, did not appear genetically programmed but related instead to subtle environmental changes.
Figures
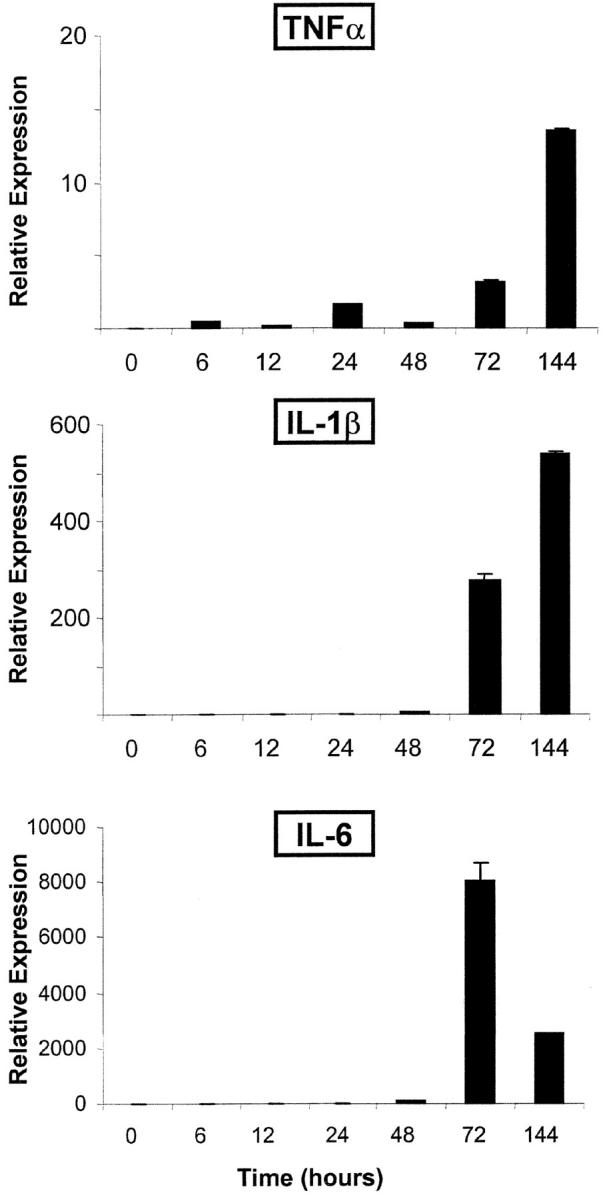
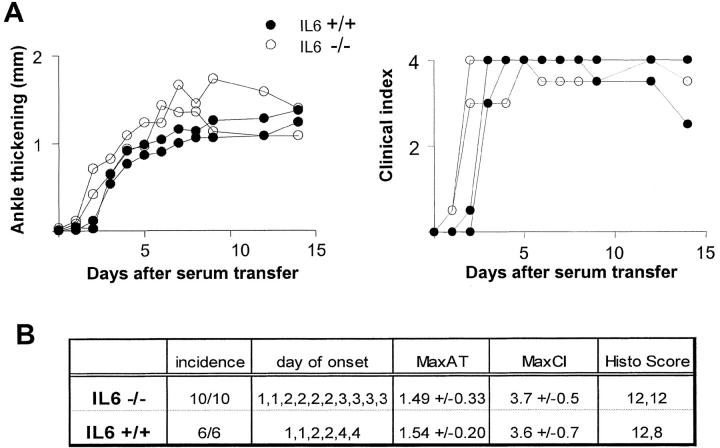
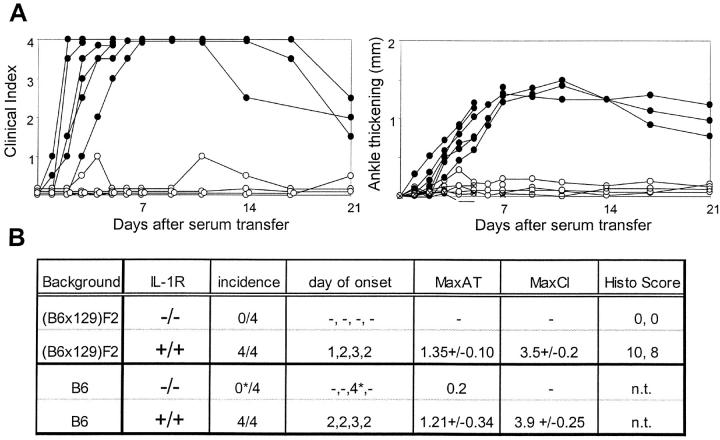
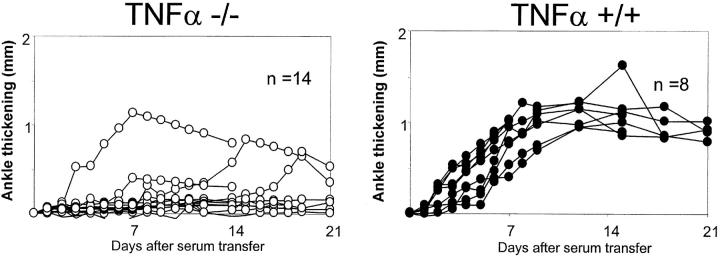
References
-
- Feldmann, M., and R.N. Maini. 2001. Anti-TNFα therapy of rheumatoid arthritis: what have we learned? Annu. Rev. Immunol. 19:163–196. - PubMed
-
- Firestein, G.S., and N.J. Zvaifler. 1990. How important are T cells in chronic rheumatoid synovitis? Arthritis Rheum. 33:768–773. - PubMed
-
- Kouskoff, V., A.-S. Korganow, V. Duchatelle, C. Degott, C. Benoist, and D. Mathis. 1996. Organ-specific disease provoked by systemic autoreactivity. Cell. 87:811–822. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
