

A phylogenomic approach to bacterial phylogeny: evidence of a core of genes sharing a common history
- PMID: 12097345
- PMCID: PMC186629
- DOI: 10.1101/gr.187002
A phylogenomic approach to bacterial phylogeny: evidence of a core of genes sharing a common history
Abstract
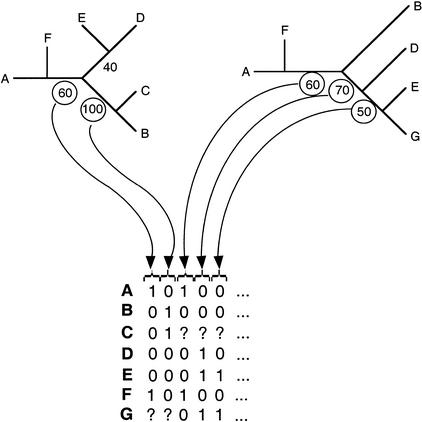
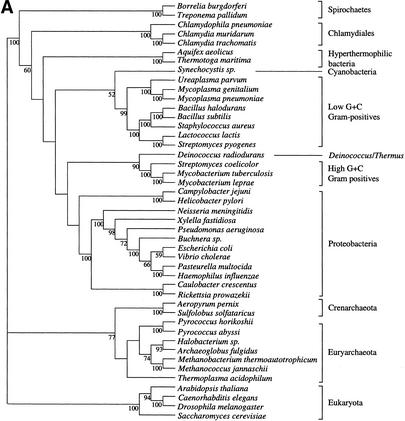
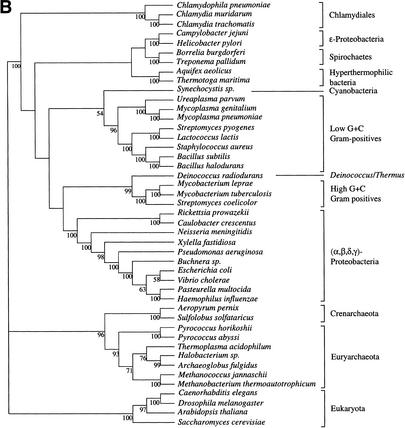
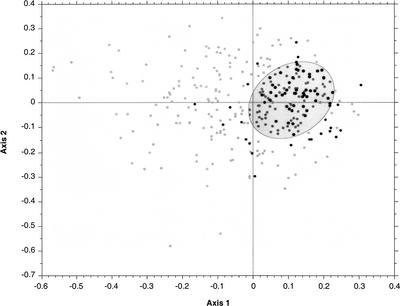
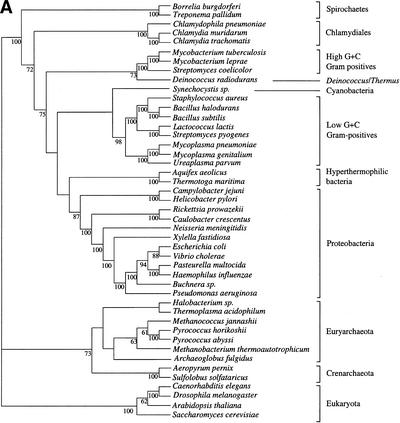
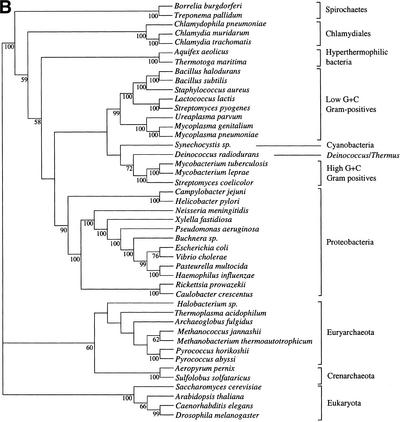
It has been claimed that complete genome sequences would clarify phylogenetic relationships between organisms, but up to now, no satisfying approach has been proposed to use efficiently these data. For instance, if the coding of presence or absence of genes in complete genomes gives interesting results, it does not take into account the phylogenetic information contained in sequences and ignores hidden paralogies by using a BLAST reciprocal best hit definition of orthology. In addition, concatenation of sequences of different genes as well as building of consensus trees only consider the few genes that are shared among all organisms. Here we present an attempt to use a supertree method to build the phylogenetic tree of 45 organisms, with special focus on bacterial phylogeny. This led us to perform a phylogenetic study of congruence of tree topologies, which allows the identification of a core of genes supporting similar species phylogeny. We then used this core of genes to infer a tree. This phylogeny presents several differences with the rRNA phylogeny, notably for the position of hyperthermophilic bacteria.
Figures
References
-
- Baldauf SL, Roger AJ, Wenk-Siefert I, Doolittle WF. A kingdom-level phylogeny of eukaryotes based on combined protein data. Science. 2000;290:972–977. - PubMed
-
- Baum BR. Combining trees as a way of combining data sets for phylogenetic inference, and the desirability of combining gene trees. Taxon. 1992;41:3–10.
-
- Bellgard MI, Itoh T, Watanabe H, Imanishi T, Gojobori T. Dynamic evolution of genomes and the concept of genome space. Ann NY Acad Sci. 1999;870:293–300. - PubMed
-
- Bocchetta M, Gribaldo S, Sanangelantoni A, Cammarano P. Phylogenetic depth of the bacterial genera Aquifex and Thermotoga inferred from analysis of ribosomal protein, elongation factor, and RNA polymerase subunit sequences. J Mol Evol. 2000;50:366–380. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Research Materials