

Expression and genomic analysis of midasin, a novel and highly conserved AAA protein distantly related to dynein
- PMID: 12102729
- PMCID: PMC117441
- DOI: 10.1186/1471-2164-3-18
Expression and genomic analysis of midasin, a novel and highly conserved AAA protein distantly related to dynein
Abstract
Background: The largest open reading frame in the Saccharomyces genome encodes midasin (MDN1p, YLR106p), an AAA ATPase of 560 kDa that is essential for cell viability. Orthologs of midasin have been identified in the genome projects for Drosophila, Arabidopsis, and Schizosaccharomyces pombe.
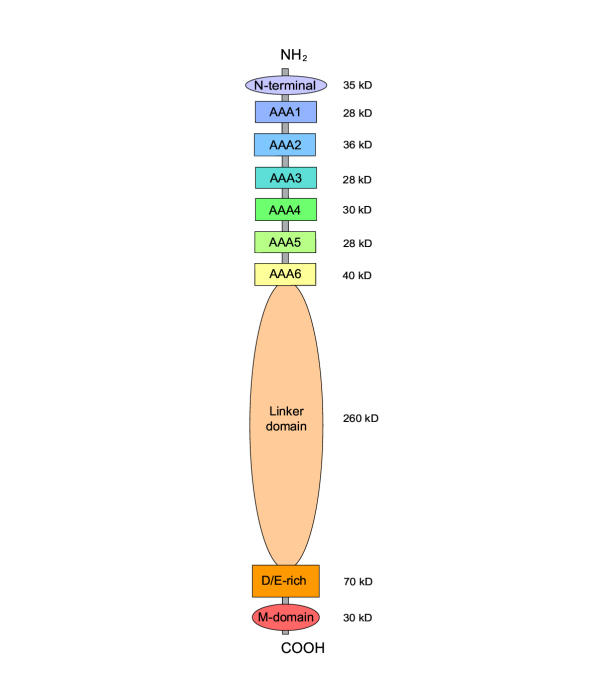
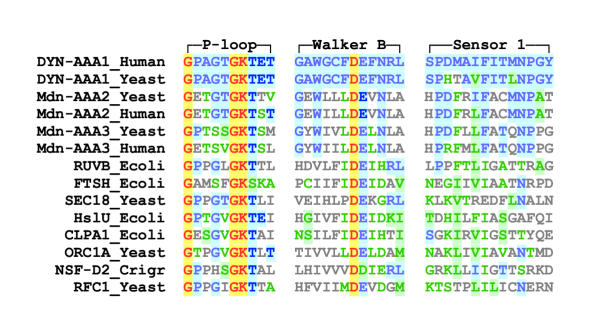
Results: Midasin is present as a single-copy gene encoding a well-conserved protein of approximately 600 kDa in all eukaryotes for which data are available. In humans, the gene maps to 6q15 and encodes a predicted protein of 5596 residues (632 kDa). Sequence alignments of midasin from humans, yeast, Giardia and Encephalitozoon indicate that its domain structure comprises an N-terminal domain (35 kDa), followed by an AAA domain containing six tandem AAA protomers (approximately 30 kDa each), a linker domain (260 kDa), an acidic domain (approximately 70 kDa) containing 35-40% aspartate and glutamate, and a carboxy-terminal M-domain (30 kDa) that possesses MIDAS sequence motifs and is homologous to the I-domain of integrins. Expression of hemagglutamin-tagged midasin in yeast demonstrates a polypeptide of the anticipated size that is localized principally in the nucleus.
Conclusions: The highly conserved structure of midasin in eukaryotes, taken in conjunction with its nuclear localization in yeast, suggests that midasin may function as a nuclear chaperone and be involved in the assembly/disassembly of macromolecular complexes in the nucleus. The AAA domain of midasin is evolutionarily related to that of dynein, but it appears to lack a microtubule-binding site.
Figures








References
-
- Bassler J. et al. Identification of a 60S preribosomal particle that is closely linked to nuclear export. Mol Cell. 2001;3:517–529. - PubMed
-
- Neuwald AF, Aravind L, Spouge JL, Koonin EV. AAA+: A class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Res. 1999;3:27–43. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
