

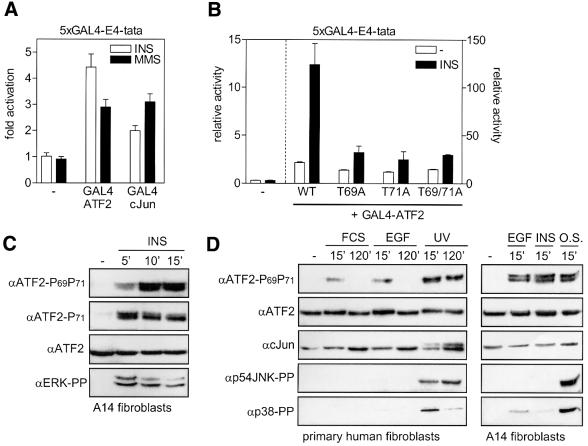
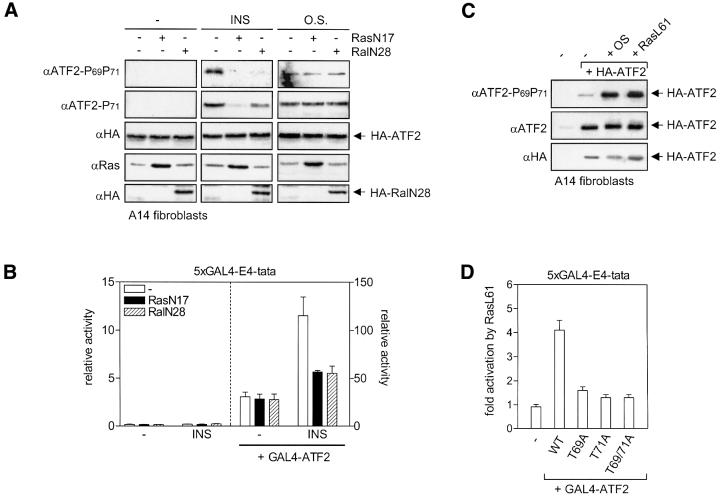
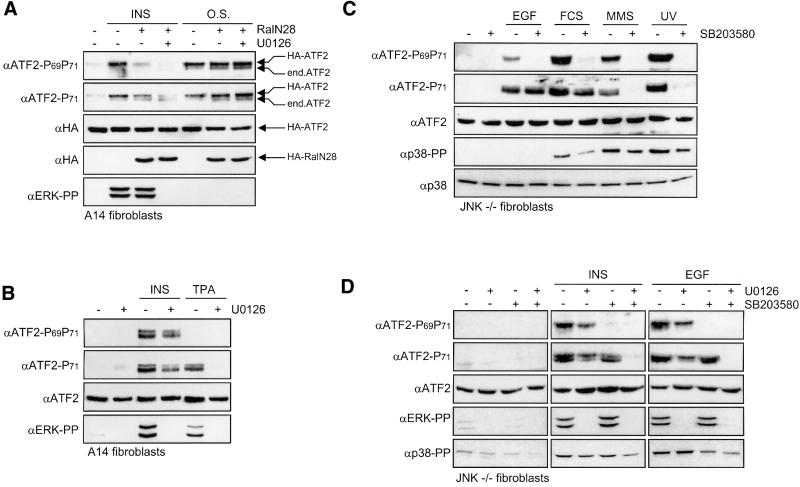
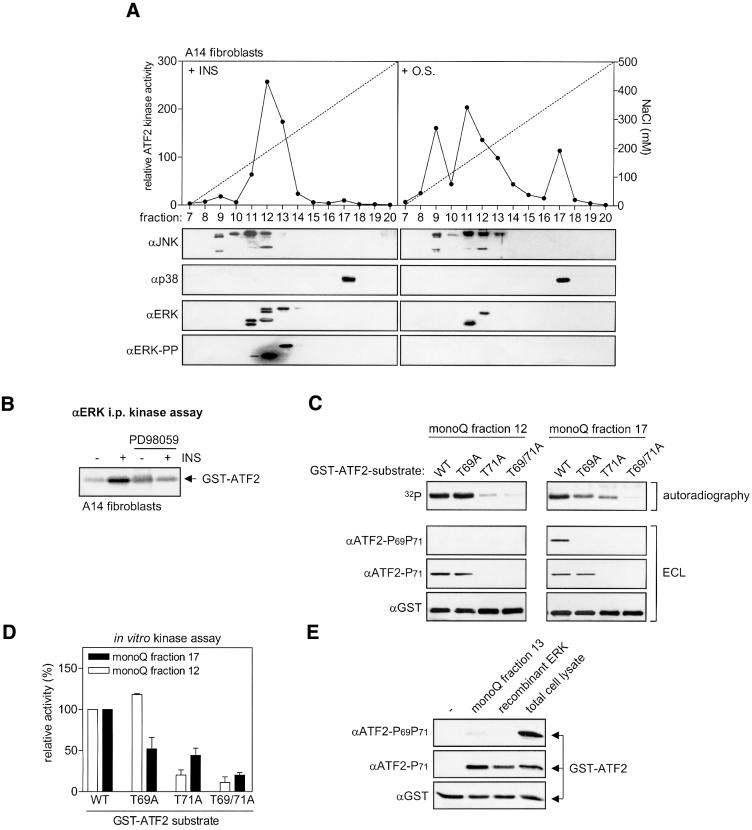
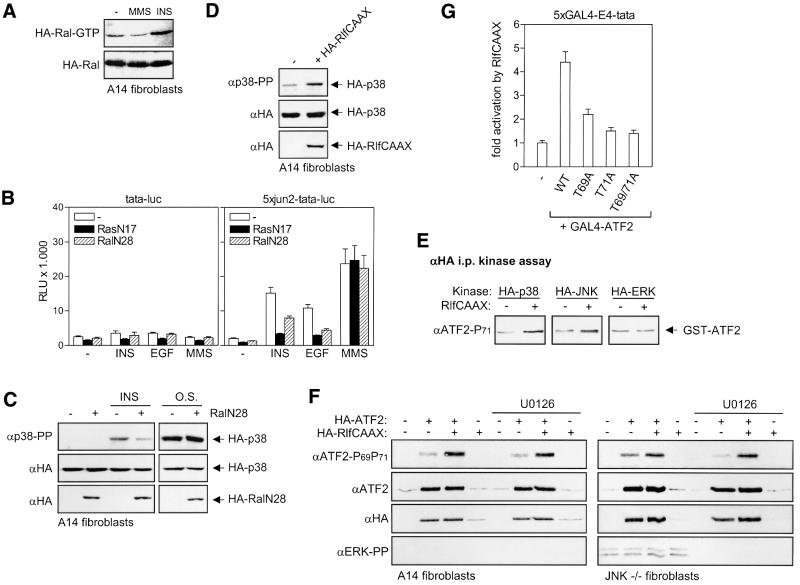
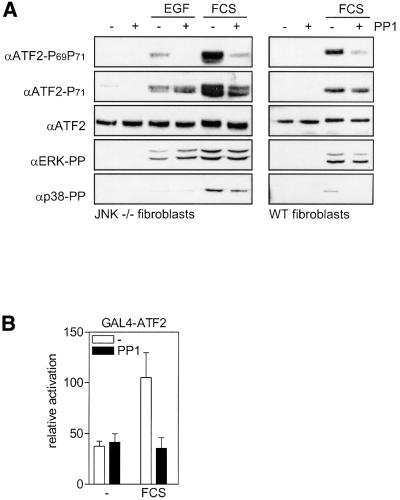
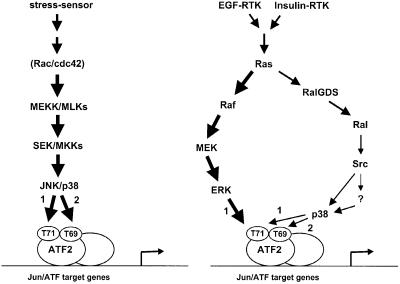
Growth factors can activate ATF2 via a two-step mechanism: phosphorylation of Thr71 through the Ras-MEK-ERK pathway and of Thr69 through RalGDS-Src-p38
- PMID: 12110590
- PMCID: PMC126107
- DOI: 10.1093/emboj/cdf361
Growth factors can activate ATF2 via a two-step mechanism: phosphorylation of Thr71 through the Ras-MEK-ERK pathway and of Thr69 through RalGDS-Src-p38
Abstract
Transcription factor ATF2 regulates gene expression in response to environmental changes. Upon exposure to cellular stresses, the mitogen-activated proteinkinase (MAPK) cascades including SAPK/JNK and p38 can enhance ATF2's transactivating function through phosphorylation of Thr69 and Thr71. How ever, the mechanism of ATF2 activation by growth factors that are poor activators of JNK and p38 is still elusive. Here, we show that in fibroblasts, insulin, epidermal growth factor (EGF) and serum activate ATF2 via a so far unknown two-step mechanism involving two distinct Ras effector pathways: the Raf-MEK-ERK pathway induces phosphorylation of ATF2 Thr71, whereas subsequent ATF2 Thr69 phosphorylation requires the Ral-RalGDS-Src-p38 pathway. Cooperation between ERK and p38 was found to be essential for ATF2 activation by these mitogens; the activity of p38 and JNK/SAPK in growth factor-stimulated fibroblasts is insufficient to phosphorylate ATF2 Thr71 or Thr69 + 71 significantly by themselves, while ERK cannot dual phosphorylate ATF2 Thr69 + 71 efficiently. These results reveal a so far unknown mechanism by which distinct MAPK pathways and Ras effector pathways cooperate to activate a transcription factor.
Figures
Similar articles
-
The role of c-Jun N-terminal kinase, p38, and extracellular signal-regulated kinase in insulin-induced Thr69 and Thr71 phosphorylation of activating transcription factor 2.Mol Endocrinol. 2006 Aug;20(8):1786-95. doi: 10.1210/me.2005-0289. Epub 2006 Apr 6. Mol Endocrinol. 2006. PMID: 16601071
-
Retinoblastoma protein interacts with ATF2 and JNK/p38 in stimulating the transforming growth factor-beta2 promoter.Arch Biochem Biophys. 2001 Oct 1;394(1):1-12. doi: 10.1006/abbi.2001.2518. Arch Biochem Biophys. 2001. PMID: 11566021
-
ORF61 protein of Varicella-zoster virus influences JNK/SAPK and p38/MAPK phosphorylation.J Med Virol. 2005 Jul;76(3):424-33. doi: 10.1002/jmv.20373. J Med Virol. 2005. PMID: 15902710
-
Changes in androgen receptor nongenotropic signaling correlate with transition of LNCaP cells to androgen independence.Cancer Res. 2004 Oct 1;64(19):7156-68. doi: 10.1158/0008-5472.CAN-04-1121. Cancer Res. 2004. PMID: 15466214
-
ATF2 on the double - activating transcription factor and DNA damage response protein.Pigment Cell Res. 2007 Dec;20(6):498-506. doi: 10.1111/j.1600-0749.2007.00414.x. Pigment Cell Res. 2007. PMID: 17935492 Free PMC article. Review.
Cited by
-
Rit GTPase regulates a p38 MAPK-dependent neuronal survival pathway.Neurosci Lett. 2012 Dec 7;531(2):125-30. doi: 10.1016/j.neulet.2012.10.036. Epub 2012 Nov 2. Neurosci Lett. 2012. PMID: 23123784 Free PMC article.
-
Identification of a novel amino acid response pathway triggering ATF2 phosphorylation in mammals.Mol Cell Biol. 2009 Dec;29(24):6515-26. doi: 10.1128/MCB.00489-09. Epub 2009 Oct 12. Mol Cell Biol. 2009. PMID: 19822663 Free PMC article.
-
c-Jun NH(2)-terminal kinase is essential for the regulation of AP-1 by tumor necrosis factor.Mol Cell Biol. 2003 Apr;23(8):2871-82. doi: 10.1128/MCB.23.8.2871-2882.2003. Mol Cell Biol. 2003. PMID: 12665585 Free PMC article.
-
Transcriptional switch by activating transcription factor 2-derived peptide sensitizes melanoma cells to apoptosis and inhibits their tumorigenicity.Proc Natl Acad Sci U S A. 2004 Mar 23;101(12):4222-7. doi: 10.1073/pnas.0400195101. Epub 2004 Mar 9. Proc Natl Acad Sci U S A. 2004. PMID: 15010535 Free PMC article.
-
Isoform-specific optical activation of kinase function reveals p38-ERK signaling crosstalk.RSC Chem Biol. 2023 Aug 25;4(10):765-773. doi: 10.1039/d2cb00157h. eCollection 2023 Oct 4. RSC Chem Biol. 2023. PMID: 37799579 Free PMC article.
References
-
- Angel P., Hattori,K., Smeal,T. and Karin,M. (1988) The jun proto-oncogene is positively autoregulated by its product, Jun/AP-1. Cell, 55, 875–885. - PubMed
-
- Bar-Sagi D. and Hall,A. (2000) Ras and Rho GTPases: a family reunion. Cell, 103, 227–238. - PubMed
-
- Beier F., Taylor,A.C. and LuValle,P. (2000) Activating transcription factor 2 is necessary for maximal activity and serum induction of the cyclin A promoter in chondrocytes. J. Biol. Chem., 275, 12948–12953. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
