

doi: 10.1101/gad.997502.
A nonsense mutation in the fibrillin-1 gene of a Marfan syndrome patient induces NMD and disrupts an exonic splicing enhancer
Affiliations
- PMID: 12130535
- PMCID: PMC186389
- DOI: 10.1101/gad.997502
Item in Clipboard
A nonsense mutation in the fibrillin-1 gene of a Marfan syndrome patient induces NMD and disrupts an exonic splicing enhancer
Genes Dev.
.
Abstract
A nonsense mutation in the fibrillin-1 (FBN1) gene of a Marfan syndrome (MFS) patient induces in-frame exon skipping of FBN1 exon 51. We present evidence, based on both in vivo and in vitro experiments, that the skipping of this exon is due to the disruption of an SC35-dependent splicing enhancer within exon 51. In addition, this nonsense mutation induces nonsense-mediated decay (NMD), which degrades the normally spliced mRNA in the patient's cells. In contrast to NMD, skipping of FBN1 exon 51 does not require translation.
Figures
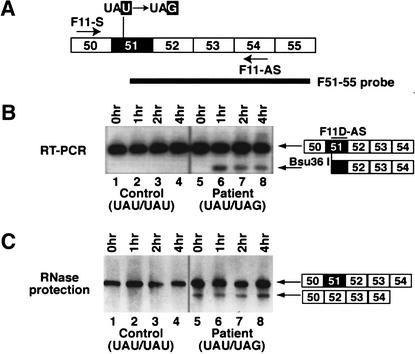
FBN1 mRNAs carrying the Y2113X nonsense mutation are subject to NMD. (A) Schematic of the FBN1 exon 51 region. The Y2113X nonsense mutation is indicated, the primers used in the RT–PCR assay are indicated by arrows, and the probe utilized in the RNase protection assay is indicated by the heavy black line. (B) RT–PCR analysis of FBN1 transcripts in the presence of anisomycin, an inhibitor of protein synthesis. Fibroblasts from an MFS patient, harboring the mutant allele causing exon skipping (lanes 5–8), and from a healthy individual as control (lanes 1–4) were exposed to 100 μg/mL anisomycin for the indicated times (0 h, untreated). RT–PCR amplification was carried out with primers F11S and F11-AS. The amplified DNA was cleaved with Bsu36 I, which cleaves at the site of the nonsense mutation. The DNA products were separated by electrophoresis, Southern blotted, and hybridized with the oligonucleotide probe F11D-AS. (C) RNase protection analysis shows that exon skipping does not require protein synthesis. RNA isolated from patient and control fibroblasts after exposure to anisomycin was subjected to RNase protection analysis with the F51–55 probe shown in A.

Mutations causing exon skipping in vivo. (A) Schematic of the OAT-FBN chimeric minigene used in this study. Mutations introduced in exon 51 are indicated below the sequence. (*) The site of the PTC at +26, which caused exon skipping in an MFS patient. (B) RT–PCR analysis of transcripts of the OAT-FBN minigenes, transiently transfected into human fibroblasts. Exon 51 skipping and inclusion was assayed with the wild-type construct (lane 2) and with constructs carrying PTCs at different positions: codon 2113 (lane 3), upstream (US) (lane 5), and downstream (DS) (lane 6). RNAs with missense mutations in a polypurine element (PPE) were assayed in lane 4. In lane 1, cells were mock transfected as a control.

In vitro splicing of dsx-FBN substrates. (A) Schematic representation of dsx-FBN splicing substrates. Dsx exonic sequences (dark boxes), ASLV and exon 6D sequences (light boxes) are shown. PTC PPE and US-PTC mutations indicated here correspond to the mutations in Figure 2A. (B) In vitro splicing reactions. The indicated radiolabeled pre-mRNAs were incubated in in vitro splicing reactions containing HeLa cell nuclear extract. RNA precursors and spliced products are indicated by the schematics at the left of the autoradiogram.

In vitro splicing of the dsx-FBN WT substrate in HeLa S100 extract complemented with SR proteins. The dsxFBN WT radiolabeled pre-mRNA was incubated in in vitro splicing reactions containing HeLa S100 extract (lanes 2–7 and 9–14) or HeLa nuclear extracts (lanes 1,8). Roughly 200 ng of each SR protein, purified from calf thymus, or 500 ng of a total SR protein preparation from HeLa cells was added to the splicing mixture as indicated.
Comment in
-
NASty effects on fibrillin pre-mRNA splicing: another case of ESE does it, but proposals for translation-dependent splice site choice live on.Genes Dev. 2002 Jul 15;16(14):1743-53. doi: 10.1101/gad.1014502. Genes Dev. 2002. PMID: 12130534 Review. No abstract available.
References
-
- Carter MS, Doskow J, Morris P, Li S, Nhim RP, Sandstedt S, Wilkinson MF. A regulatory mechanism that detects premature nonsense codons in T-cell receptor transcripts in vivo is reversed by protein synthesis inhibitors in vitro. J Biol Chem. 1995;270:28995–29003. - PubMed
-
- Dietz HC, Kendzior RJ., Jr Maintenance of an open reading frame as an additional level of scrutiny during splice site selection. Nat Genet. 1994;8:183–188. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases