

The zinc finger domain of NEMO is selectively required for NF-kappa B activation by UV radiation and topoisomerase inhibitors
- PMID: 12138192
- PMCID: PMC133970
- DOI: 10.1128/MCB.22.16.5813-5825.2002
The zinc finger domain of NEMO is selectively required for NF-kappa B activation by UV radiation and topoisomerase inhibitors
Abstract
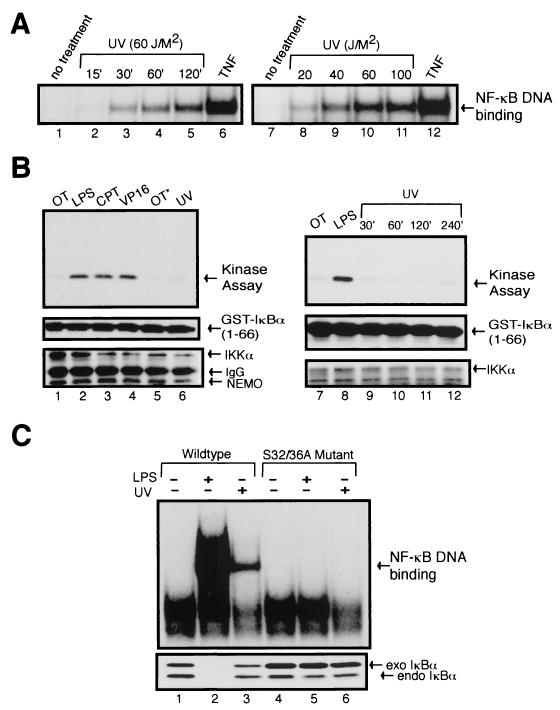
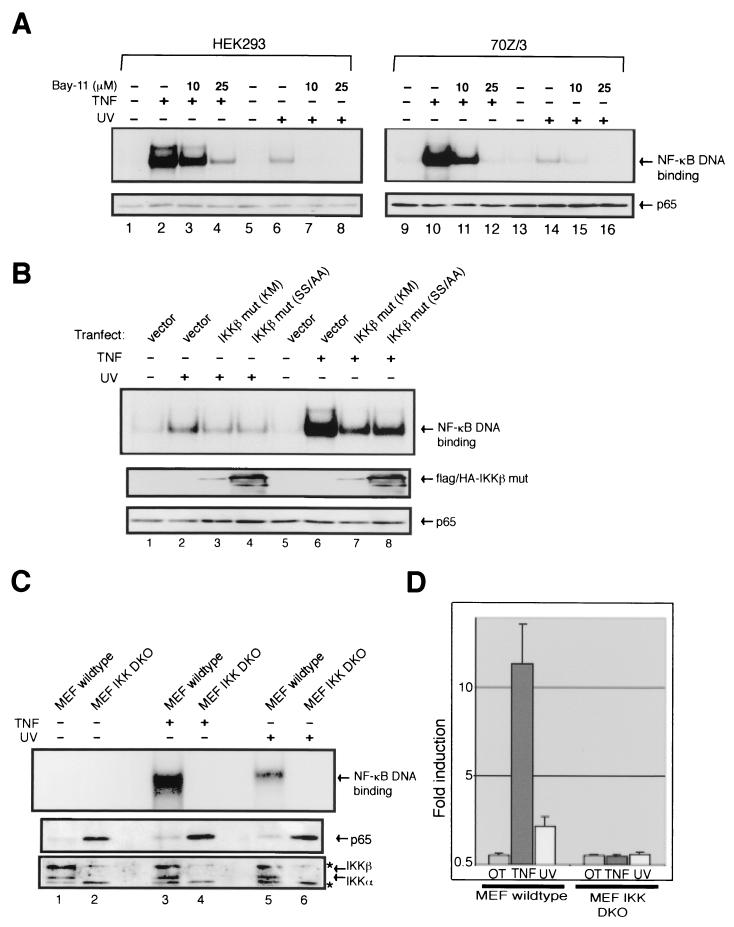
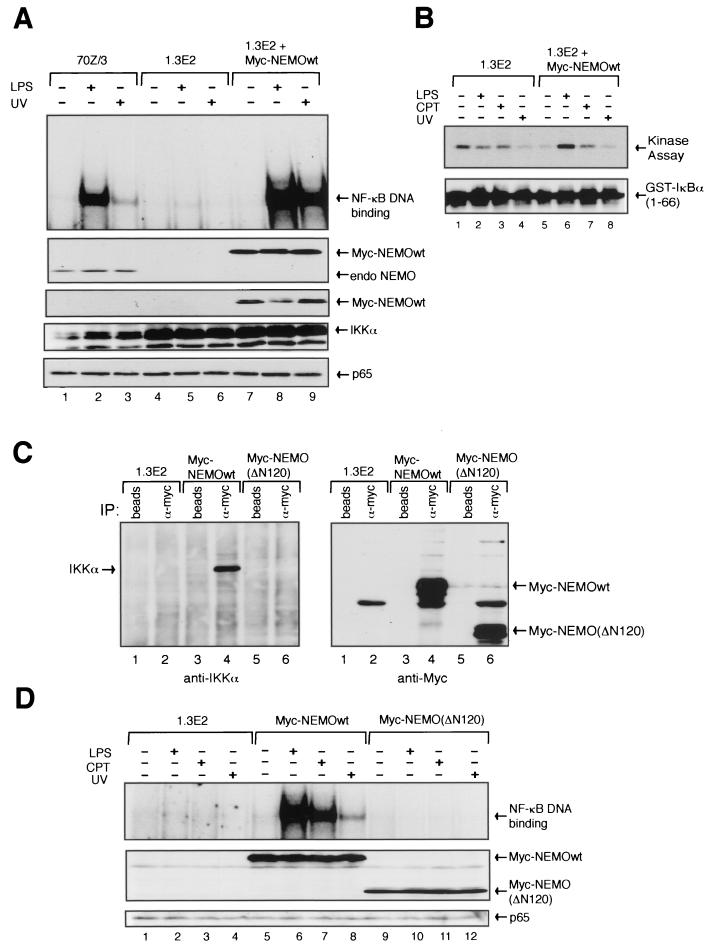
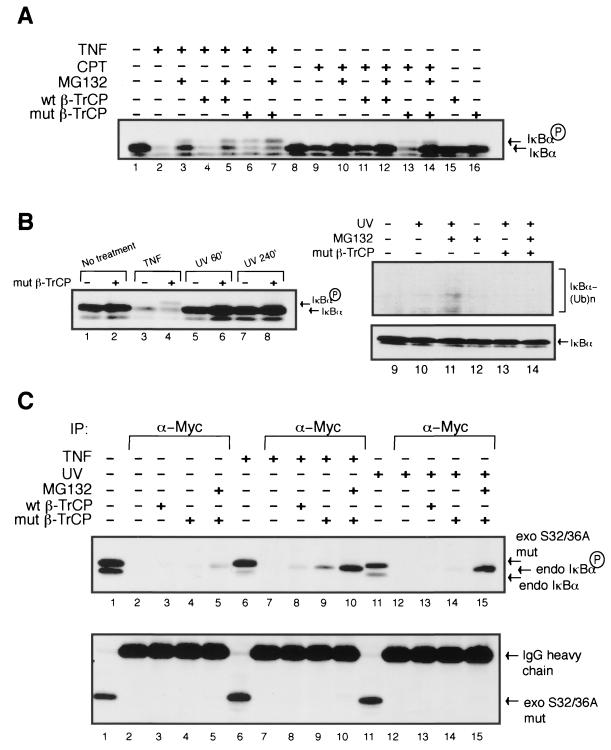
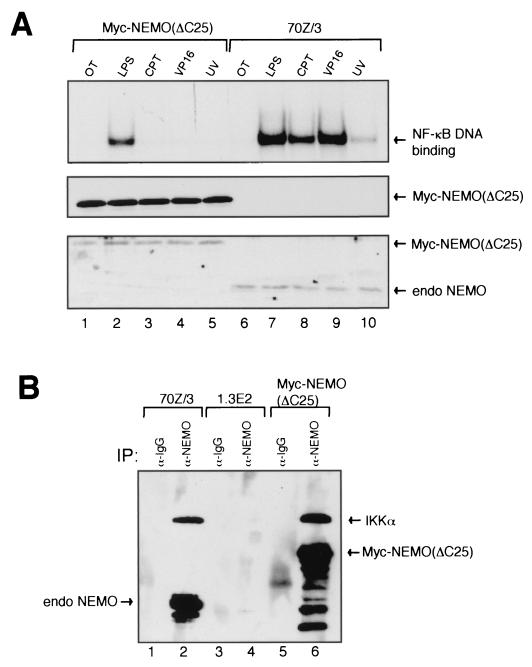
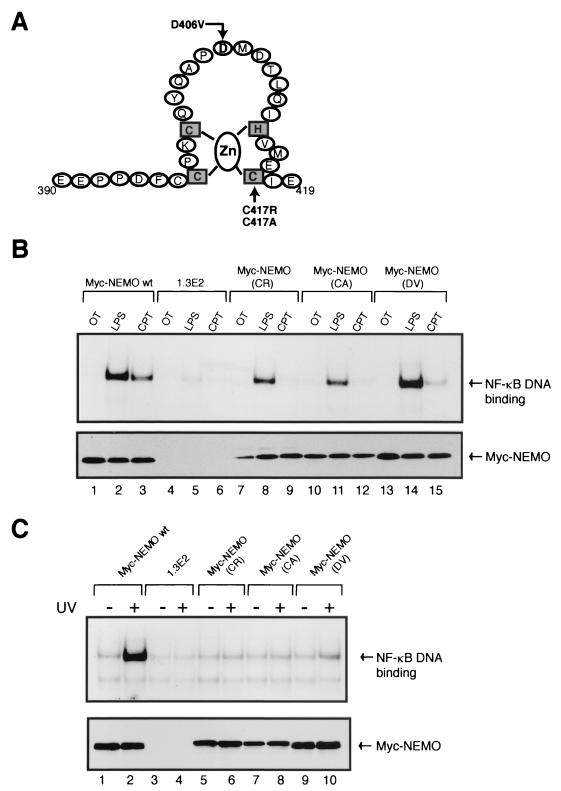
Exposure of mammalian cells to UV radiation was proposed to stimulate the transcription factor NF-kappa B by a unique mechanism. Typically, rapid and strong inducers of NF-kappa B, such as tumor necrosis factor alpha (TNF-alpha) and bacterial lipopolysaccharide (LPS), lead to rapid phosphorylation and proteasomal degradation of its inhibitory protein, I kappa B alpha. In contrast, UV, a relatively slower and weaker inducer of NF-kappa B, was suggested not to require phosphorylation of I kappa B alpha for its targeted degradation by the proteasome. We now provide evidence to account for this peculiar degradation process of I kappa B alpha. The phospho-I kappa B alpha generated by UV is only detectable by expressing a Delta F-box mutant of the ubiquitin ligase beta-TrCP, which serves as a specific substrate trap for serine 32 and 36 phosphorylated I kappa B alpha. In agreement with this finding, we also find that the I kappa B kinase (IKK) phospho-acceptor sites on I kappa B alpha, core components of the IKK signalsome, and IKK catalytic activity are all required for UV signaling. Furthermore, deletion and point mutation analyses reveal that both the amino-terminal IKK-binding and the carboxy-terminal putative zinc finger domains of NEMO (IKK gamma) are critical for UV-induced NF-kappa B activation. Interestingly, the zinc finger domain is also required for NF-kappa B activation by two other slow and weak inducers, camptothecin and etoposide. In contrast, the zinc finger module is largely dispensable for NF-kappa B activation by the rapid and strong inducers LPS and TNF-alpha. Thus, we suggest that the zinc finger domain of NEMO likely represents a point of convergence for signaling pathways initiated by slow and weak NF-kappa B-activating conditions.
Figures
References
-
- Cao, Z. D., J. Xiong, M. Takeuchi, T. Kurama, and D. V. Goeddel. 1996. TRAF6 is a signal transducer for interleukin-1. Nature 383:443-446. - PubMed
-
- Chen, Z., J. Hagler, V. J. Palombella, F. Melandri, D. Scherer, D. Ballard, and T. Maniatis. 1995. Signal-induced site-specific phosphorylation targets IκBα to the ubiquitin-proteasome pathway. Genes Dev. 9:1586-1597. - PubMed
-
- Chen, Z. J., L. Parent, and T. Maniatis. 1996. Site-specific phosphorylation of IκBα by a novel ubiquitination-dependent protein kinase activity. Cell 84:853-862. - PubMed
-
- Delhase, M., M. Hayakawa, Y. Chen, and M. Karin. 1999. Positive and negative regulation of IκB kinase activity through IKKβ subunit phosphorylation. Science 284:309-313. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous