

Energetic and entropic contributions to the interactions between like-charged groups in cationic peptides: A molecular dynamics simulation study
- PMID: 12142454
- PMCID: PMC2373690
- DOI: 10.1110/ps.0203002
Energetic and entropic contributions to the interactions between like-charged groups in cationic peptides: A molecular dynamics simulation study
Abstract
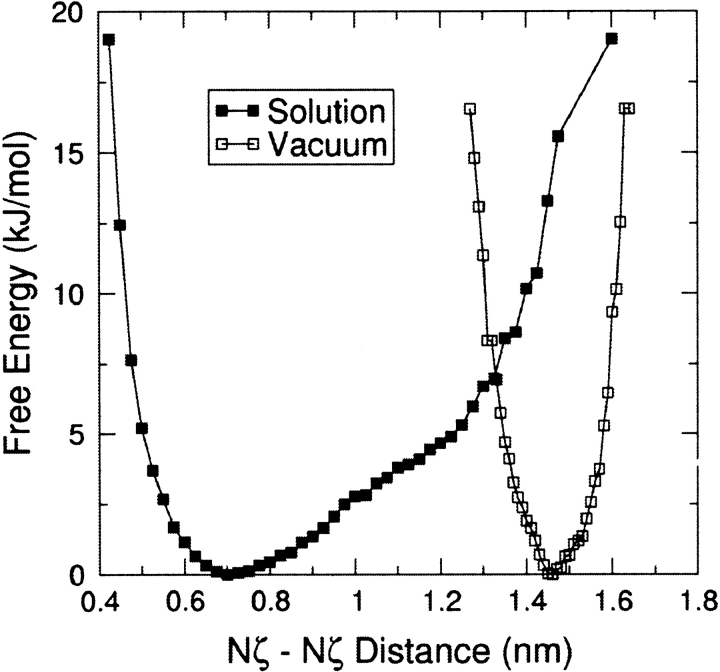
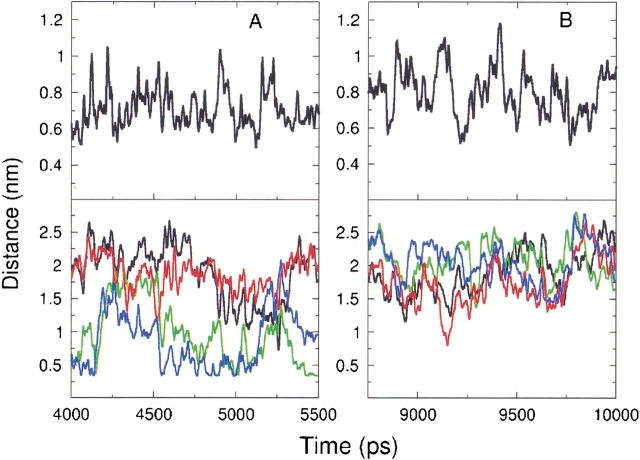
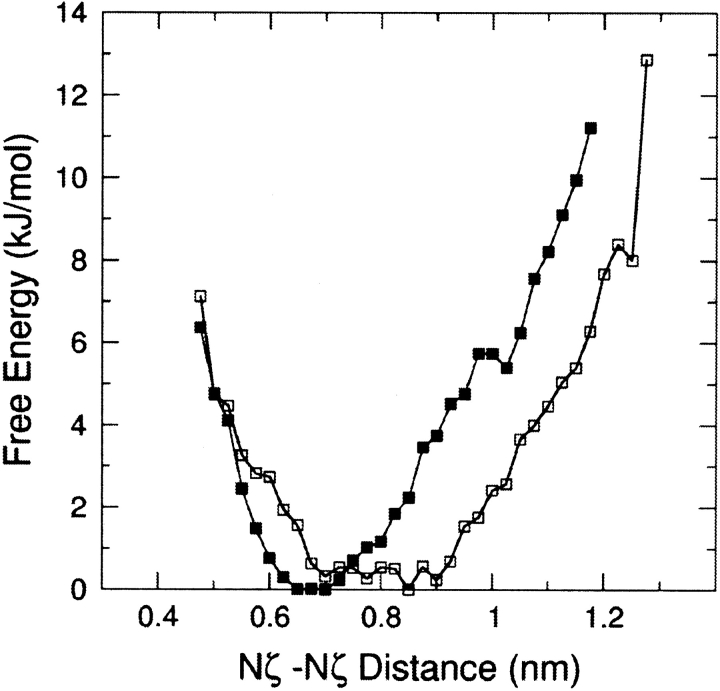
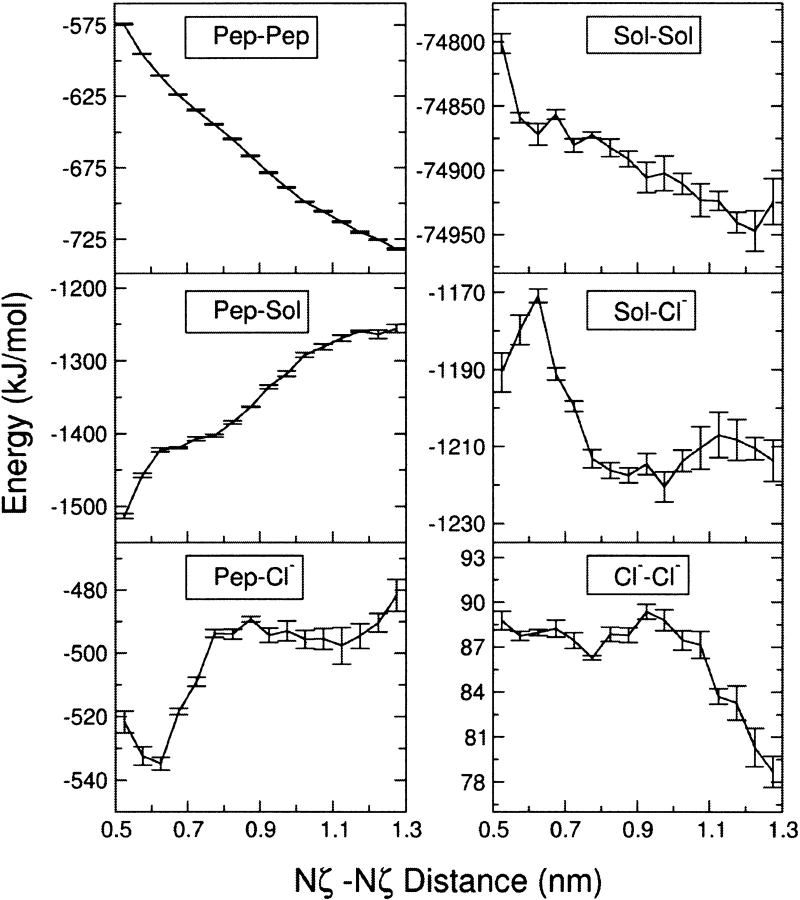
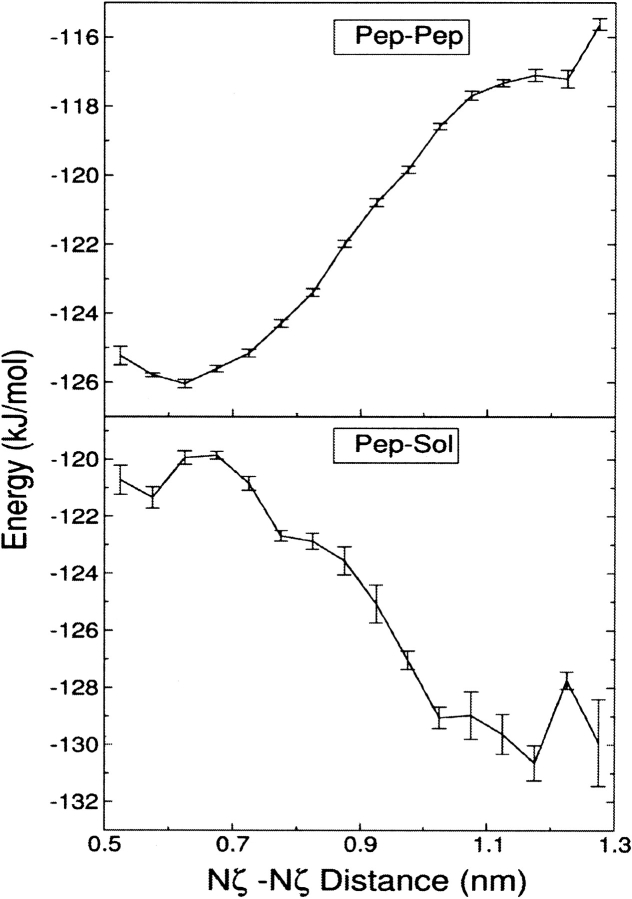
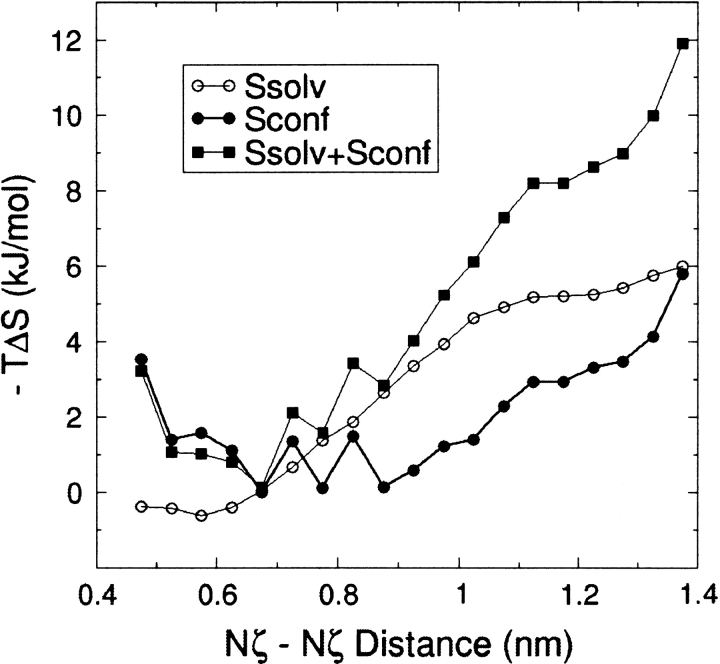
The interaction between like-charged amino acid residues has been proposed to stabilize the folded state of peptides and proteins, and to modulate the substrate binding and the action mechanism of enzymes. We have used an alanine- and lysine-based peptide as a model system to study the interaction between like charges, and we have performed a 16-nsec molecular dynamics simulation in solution. The calculated potential of mean force for the approach of the lysine's Nzeta atoms showed a minimum at a distance of 0.7 nm, in agreement with the separation probabilities obtained from analysis of protein crystal structures. The analysis of the individual energy components showed that the solvent polarization pays for the approach of the like charges and that the van der Waals energies do not contribute significantly. The entropic contributions have been divided in conformational and desolvation terms. Both terms favor the formation of the charge pair. A 10-fold increase in counterion concentration was observed-with respect to its bulk concentration-next to the peptide charges, which helps to stabilize the peptide charges at a close distance.
Figures

Similar articles
-
ES/IS: estimation of conformational free energy by combining dynamics simulations with explicit solvent with an implicit solvent continuum model.Biophys Chem. 1999 Apr 5;78(1-2):195-205. doi: 10.1016/s0301-4622(98)00230-0. Biophys Chem. 1999. PMID: 10343388
-
Molecular dynamics simulation of folding of a short helical peptide with many charged residues.J Phys Chem B. 2005 Oct 27;109(42):19980-6. doi: 10.1021/jp052349k. J Phys Chem B. 2005. PMID: 16853583
-
Association of helical beta-peptides and their aggregation behavior from the potential of mean force in explicit solvent.Biophys J. 2009 Jun 3;96(11):4349-62. doi: 10.1016/j.bpj.2008.11.076. Biophys J. 2009. PMID: 19486660 Free PMC article.
-
Peptide folding driven by Van der Waals interactions.Protein Sci. 2015 Sep;24(9):1383-8. doi: 10.1002/pro.2710. Epub 2015 Jun 11. Protein Sci. 2015. PMID: 26013298 Free PMC article.
-
Protein stabilization by salt bridges: concepts, experimental approaches and clarification of some misunderstandings.J Mol Recognit. 2004 Jan-Feb;17(1):1-16. doi: 10.1002/jmr.657. J Mol Recognit. 2004. PMID: 14872533 Review.
Cited by
-
Simple physics-based analytical formulas for the potentials of mean force of the interaction of amino-acid side chains in water. V. Like-charged side chains.J Phys Chem B. 2011 May 19;115(19):6119-29. doi: 10.1021/jp111258p. Epub 2011 Apr 18. J Phys Chem B. 2011. PMID: 21500792 Free PMC article.
-
The Role of Oxidation Pattern and Water Content in the Spatial Arrangement and Dynamics of Oxidized Graphene-Based Aqueous Dispersions.Int J Mol Sci. 2022 Nov 3;23(21):13459. doi: 10.3390/ijms232113459. Int J Mol Sci. 2022. PMID: 36362261 Free PMC article.
-
Simple Physics-Based Analytical Formulas for the Potentials of Mean Force of the Interaction of Amino Acid Side Chains in Water. VII. Charged-Hydrophobic/Polar and Polar-Hydrophobic/Polar Side Chains.J Phys Chem B. 2017 Jan 19;121(2):379-390. doi: 10.1021/acs.jpcb.6b08541. Epub 2017 Jan 5. J Phys Chem B. 2017. PMID: 28000446 Free PMC article.
References
-
- Bader, J.S. and Chandler, D. 1992. Computer simulation study of the mean force between ferrous and ferric ions in water. J. Phys. Chem. 96 6423–6427.
-
- Barlow D.J. and Thornton, J. M. 1983. Ion-pairs in proteins. J. Mol. Biol. 168 867–885. - PubMed
-
- Berendsen, H.J.C., Postma, J.P.M., DiNola, A., and Haak, J.R. 1984. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81 3684–3690.
-
- Berendsen, H.J.C., Grigera, J.R., and Straatsma, T.P. 1987. The missing term in effective pair potentials. J. Phys. Chem. 91 6269–6271.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
