

Infantile-onset ascending hereditary spastic paralysis is associated with mutations in the alsin gene
- PMID: 12145748
- PMCID: PMC379189
- DOI: 10.1086/342359
Infantile-onset ascending hereditary spastic paralysis is associated with mutations in the alsin gene
Abstract
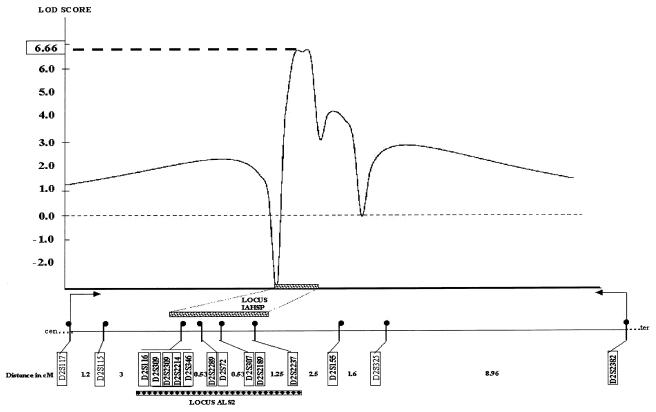
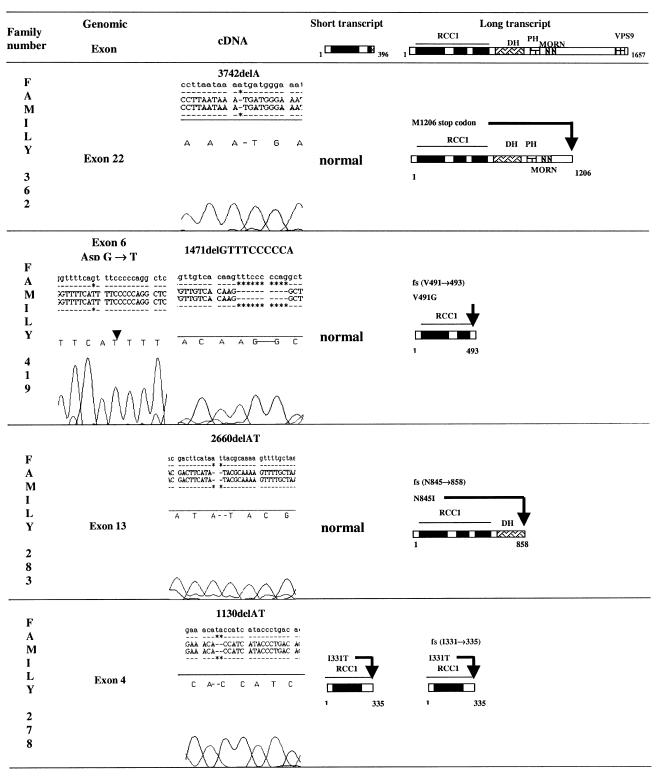
We studied 15 patients, from 10 families, who presented with severe spastic paralysis with an infantile onset and an ascending progression. Spastic paraplegia began during the first 2 years of life and extended to upper limbs within the next few years. During the first decade of life, the disease progressed to tetraplegia, anarthria, dysphagia, and slow eye movements. Overall, the disease was compatible with long survival. Signs of lower motor-neuron involvement were never observed, whereas motor-evoked potentials and magnetic resonance imaging demonstrated a primitive, pure degeneration of the upper motor neurons. Genotyping and linkage analyses demonstrated that this infantile-onset ascending hereditary spastic paralysis (IAHSP) is allelic to the condition previously reported as juvenile amyotrophic lateral sclerosis at the ALS2 locus on chromosome 2q33-35 (LOD score 6.66 at recombination fraction 0). We analyzed ALS2, recently found mutated in consanguineous Arabic families presenting either an ALS2 phenotype or juvenile-onset primary lateral sclerosis (JPLS), as a candidate gene. In 4 of the 10 families, we found abnormalities: three deletions and one splice-site mutation. All the mutations lead to a truncated alsin protein. In one case, the mutation affected both the short and the long alsin transcript. In the six remaining families, absence of cDNA ALS2 mutations suggests either mutations in regulatory ALS2 regions or genetic heterogeneity, as already reported in JPLS. Alsin mutations are responsible for a primitive, retrograde degeneration of the upper motor neurons of the pyramidal tracts, leading to a clinical continuum from infantile (IAHSP) to juvenile forms with (ALS2) or without (JPLS) lower motor-neuron involvement. Further analyses will determine whether other hereditary disorders with primitive involvement of the central motor pathways, as pure forms of spastic paraplegia, could be due to alsin dysfunction.
Figures

References
Electronic-Database Information
-
- Center for Medical Genetics, Marshfield Medical Research Foundation, http://www.marshfieldclinic.org/research/genetics/ (for order of and distance between markers)
-
- Centre de Ressources INFOBIOGEN, http://www.infobiogen.fr/ (for linkage analysis support)
-
- Généthon, http://www.genethon.fr/php/index.php (for primer sequences)
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for spastin [MIM 604277], paraplegin [MIM 604277], L1CAM [MIM 308840], PLP [MIM 312080], atlastin [MIM 606439], HSP90 [MIM 118190], SOD1 [MIM 147450], alsin [MIM 606352], SPG1 [MIM 312900], SPG2 [MIM 312920], SPG3A [MIM182600], SPG4 [MIM182601], SPG7 [MIM 602783], SPG13 [MIM 605280], ALS1 [MIM105400], ALS2 [MIM 205100], ALS4 [MIM 602433], ALS5 [MIM 602099], ALS6 [MIM 606640], and PLSJ [MIM 606353])
-
- Web Resources of Genetic Linkage Analysis, http://linkage.rockefeller.edu/ (for LINKAGE package, version 5.1, generation of pedigree file [MAKEPED] and data file [PREPLINK], using the allele frequency of each marker found in CEPH database)
References
-
- Ben Hamida M, Hentati F, Ben Hamida C (1990) Hereditary motor system diseases (chronic juvenile amyotrophic lateral sclerosis). Brain 113:347–363 - PubMed
-
- Casari G, De Fusco M, Ciarmatori S, Zeviani M, Mora M, Fernandez P, De Michele G, Filla A, Cocozza S, Marconi R, Durr A, Fontaine B, Ballabio A (1998) Spastic paraplegia and OXPHOS impairment caused by mutations in paraplegin, a nuclear-encoded mitochondrial metalloprotease. Cell 93:973–983 - PubMed
-
- Figlewicz DA, Bird TD (1999) “Pure” hereditary spastic paraplegias: the story becomes complicated. Neurology 53:5–7 - PubMed
-
- Hadano S, Hand C, Osuga H, Yanagisawa Y, Otomo A, Devon RS, Miyamoto N, Showguchi-Miyata J, Okada Y, Singaraja R, Figlewicz DA, Kwiatkowski T, Hosler BA, Sagie T, Skaug J, Nasir J, Brown RH Jr, Scherer SW, Rouleau GA, Hayden MR, Ikeda JE (2001a) A gene encoding a putative GTPase regulator is mutated in familial amyotrophic lateral sclerosis 2. Nat Genet 29:166–173 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
