

Evidence of a critical architectural function for the RAG proteins in end processing, protection, and joining in V(D)J recombination
- PMID: 12154124
- PMCID: PMC186421
- DOI: 10.1101/gad.984502
Evidence of a critical architectural function for the RAG proteins in end processing, protection, and joining in V(D)J recombination
Abstract
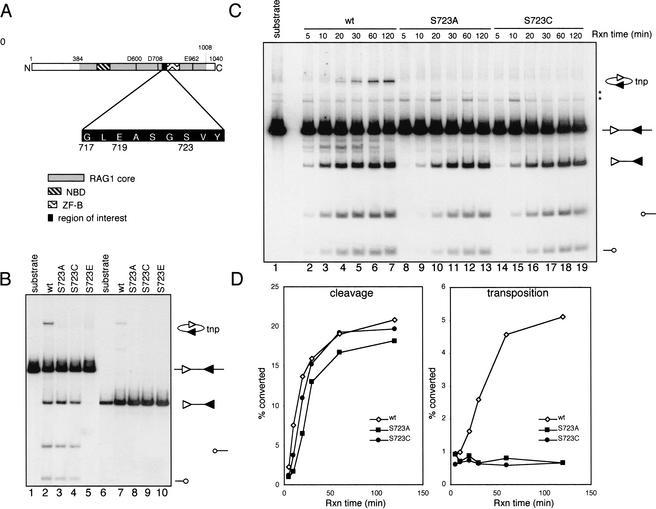
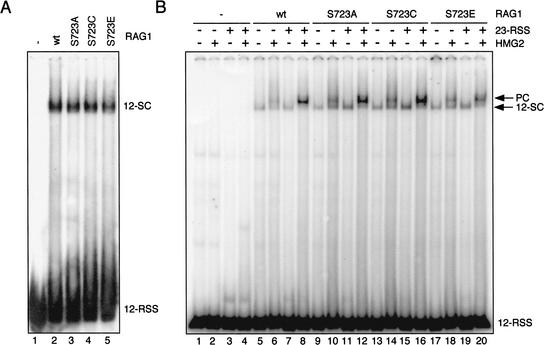
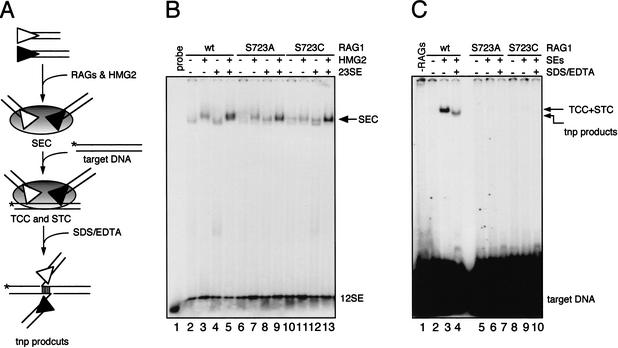
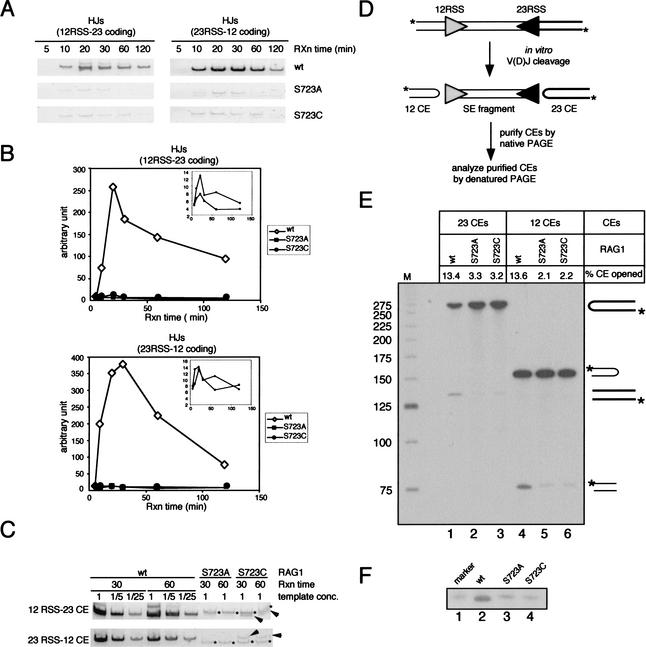
In addition to creating the DNA double strand breaks that initiate V(D)J recombination, the RAG proteins are thought to play a critical role in the joining phase of the reaction. One such role, suggested by in vitro studies, might be to ensure the structural integrity of postcleavage complexes, but the significance of such a function in vivo is unknown. We have identified RAG1 mutants that are proficient in DNA cleavage but defective in their ability to interact with coding ends after cleavage and in the capture of target DNA for transposition. As a result, these mutants exhibit severe defects in hybrid joint formation, hairpin coding end opening, and transposition in vitro, and in V(D)J recombination in vivo. Our results suggest that the RAG proteins have an architectural function in facilitating proper and efficient V(D)J joining, and a protective function in preventing degradation of broken ends prior to joining.
Figures


Similar articles
-
Mutational analysis of RAG1 and RAG2 identifies three catalytic amino acids in RAG1 critical for both cleavage steps of V(D)J recombination.Genes Dev. 1999 Dec 1;13(23):3059-69. doi: 10.1101/gad.13.23.3059. Genes Dev. 1999. PMID: 10601032 Free PMC article.
-
Joining-deficient RAG1 mutants block V(D)J recombination in vivo and hairpin opening in vitro.Mol Cell. 2001 Jan;7(1):65-75. doi: 10.1016/s1097-2765(01)00155-1. Mol Cell. 2001. PMID: 11172712
-
Different types of V(D)J recombination and end-joining defects in DNA double-strand break repair mutant mammalian cells.Eur J Immunol. 2002 Mar;32(3):701-9. doi: 10.1002/1521-4141(200203)32:3<701::AID-IMMU701>3.0.CO;2-T. Eur J Immunol. 2002. PMID: 11870614
-
The RAG proteins and V(D)J recombination: complexes, ends, and transposition.Annu Rev Immunol. 2000;18:495-527. doi: 10.1146/annurev.immunol.18.1.495. Annu Rev Immunol. 2000. PMID: 10837067 Review.
-
New concepts in the regulation of an ancient reaction: transposition by RAG1/RAG2.Immunol Rev. 2004 Aug;200:261-71. doi: 10.1111/j.0105-2896.2004.00167.x. Immunol Rev. 2004. PMID: 15242411 Review.
Cited by
-
Regulation of RAG1/RAG2-mediated transposition by GTP and the C-terminal region of RAG2.EMBO J. 2003 Apr 15;22(8):1922-30. doi: 10.1093/emboj/cdg185. EMBO J. 2003. PMID: 12682024 Free PMC article.
-
Expansion of immunoglobulin-secreting cells and defects in B cell tolerance in Rag-dependent immunodeficiency.J Exp Med. 2010 Jul 5;207(7):1541-54. doi: 10.1084/jem.20091927. Epub 2010 Jun 14. J Exp Med. 2010. PMID: 20547827 Free PMC article.
-
Lack of MSH2 involvement differentiates V(D)J recombination from other non-homologous end joining events.Nucleic Acids Res. 2005 Nov 27;33(21):6733-42. doi: 10.1093/nar/gki983. Print 2005. Nucleic Acids Res. 2005. PMID: 16314305 Free PMC article.
-
RAG: a recombinase diversified.Nat Immunol. 2009 Aug;10(8):817-21. doi: 10.1038/ni.1776. Epub 2009 Jul 21. Nat Immunol. 2009. PMID: 19621044 Free PMC article. Review.
-
Compound heterozygous mutation of Rag1 leading to Omenn syndrome.PLoS One. 2015 Apr 7;10(4):e0121489. doi: 10.1371/journal.pone.0121489. eCollection 2015. PLoS One. 2015. PMID: 25849362 Free PMC article.
References
-
- Agrawal A, Eastman QM, Schatz DG. Transposition mediated by RAG1 and RAG2 and its implications for the evolution of the immune system. Nature. 1998;394:744–751. - PubMed
-
- Agrawal A, Schatz DG. RAG1 and RAG2 form a stable post-cleavage synaptic complex with DNA containing signal ends in V(D)J recombination. Cell. 1997;89:43–53. - PubMed
-
- Aidinis V, Dias DC, Gomez CA, Bhattacharyya D, Spanopoulou E, Santagata S. Definition of minimal domains of interaction within the recombination-activating genes 1 and 2 recombinase complex. J Immunol. 2000;164:5826–5832. - PubMed
-
- Arbuckle JL, Fauss LJ, Simpson R, Ptaszek LM, Rodgers KK. Identification of two topologically independent domains in RAG1 and their role in macromolecular interactions relevant to V(D)J recombination. J Biol Chem. 2001;276:37093–37101. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials