

Discovery of a new function of cyclooxygenase (COX)-2: COX-2 is a cardioprotective protein that alleviates ischemia/reperfusion injury and mediates the late phase of preconditioning
- PMID: 12160947
- PMCID: PMC3242376
- DOI: 10.1016/s0008-6363(02)00414-5
Discovery of a new function of cyclooxygenase (COX)-2: COX-2 is a cardioprotective protein that alleviates ischemia/reperfusion injury and mediates the late phase of preconditioning
Abstract
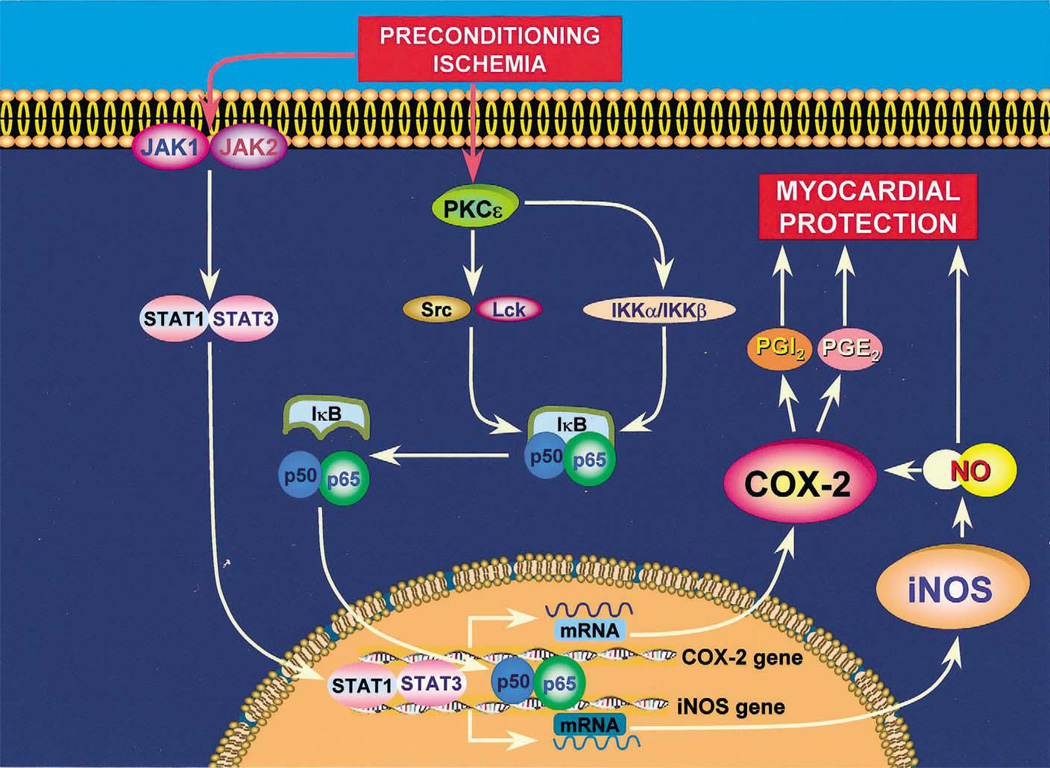
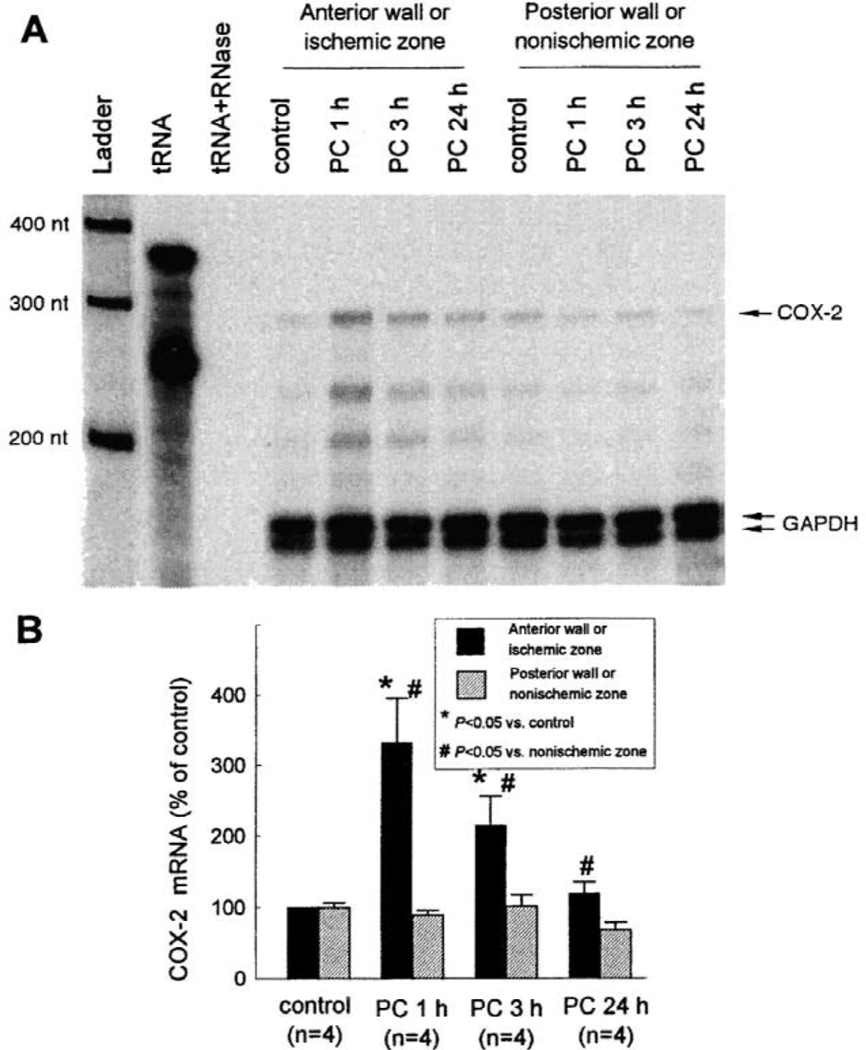
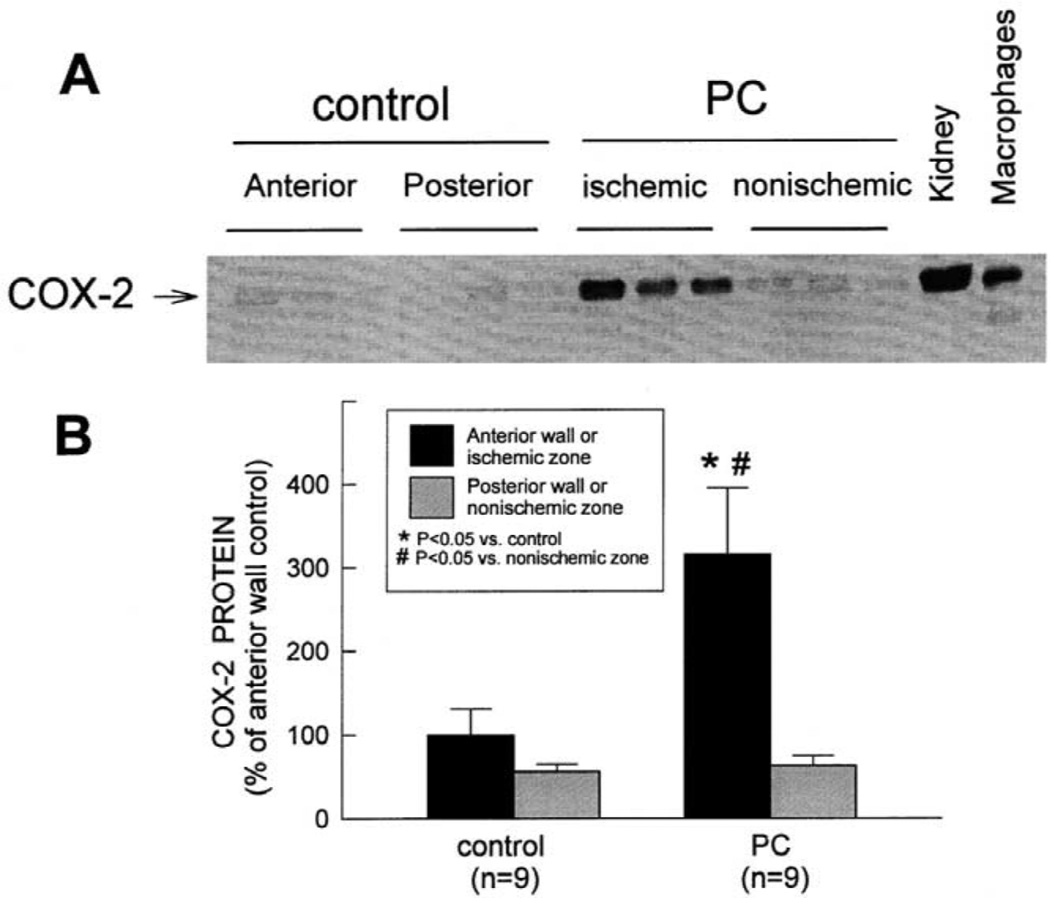
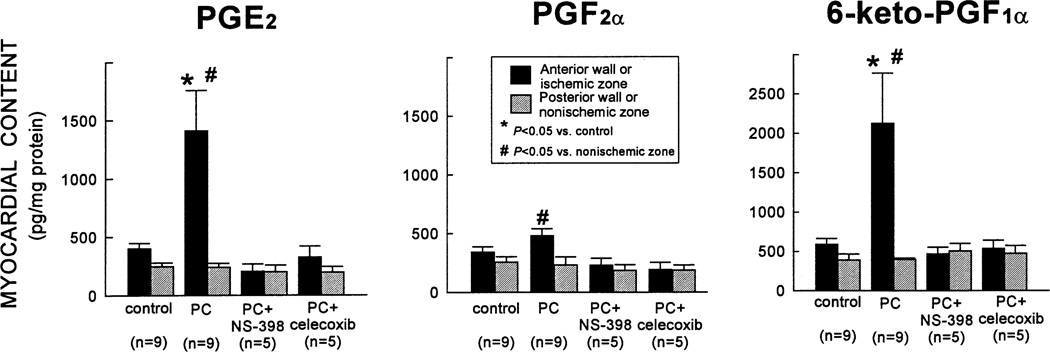
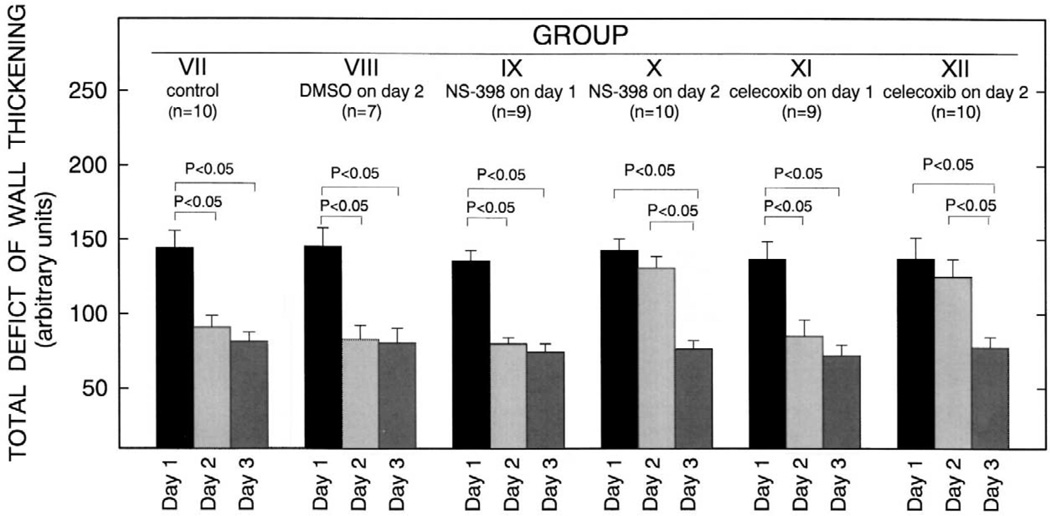
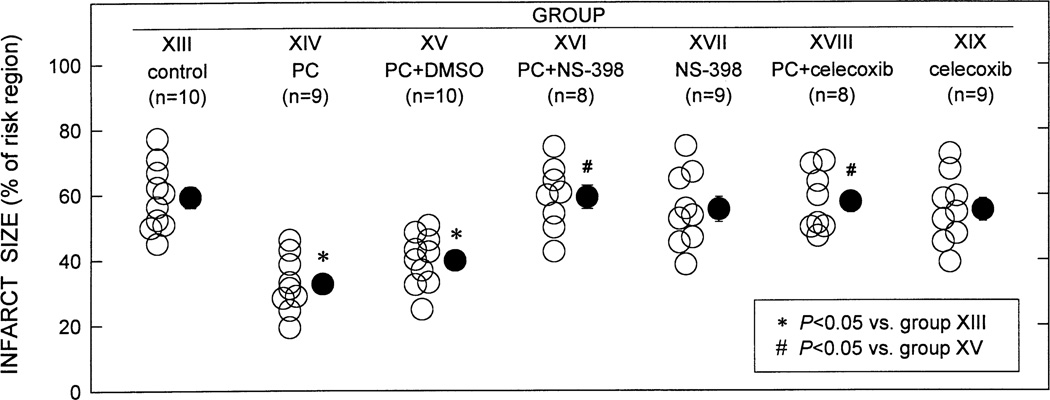
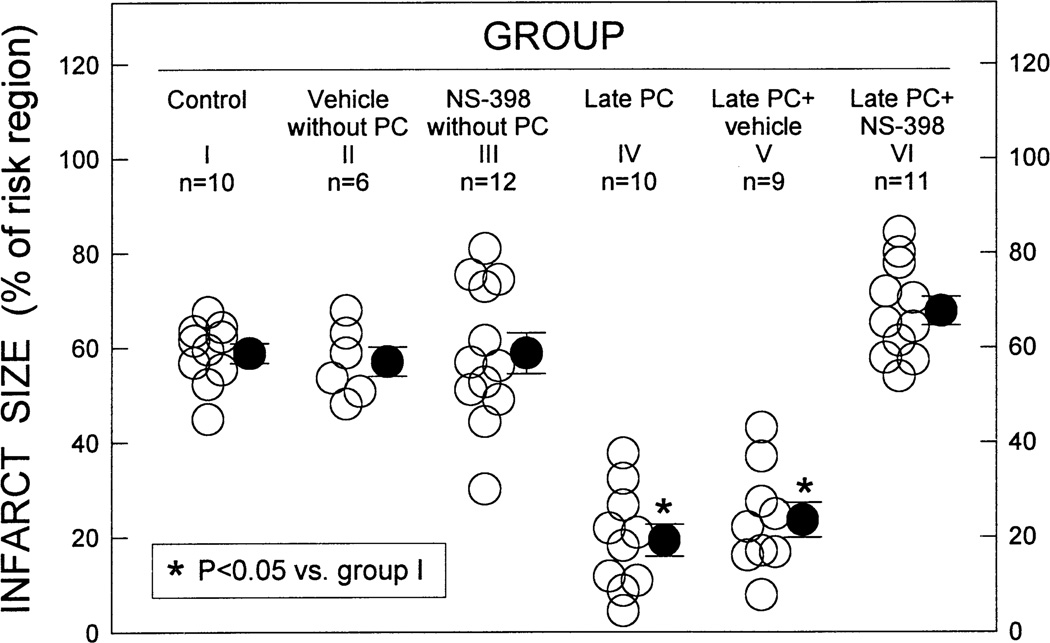
More than 10 years after its discovery, the function of cyclooxygenase-2 (COX-2) in the cardiovascular system remains largely an enigma. Many scholars have assumed that the allegedly detrimental effects of COX-2 in other systems (e.g. proinflammatory actions and tumorigenesis) signify a detrimental role of this protein in cardiovascular homeostasis as well. This view, however, is ill-founded. Recent studies have demonstrated that ischemic preconditioning (PC) upregulates the expression and activity of COX-2 in the heart, and that this increase in COX-2 activity mediates the protective effects of the late phase of PC against both myocardial stunning and myocardial infarction. An obligatory role of COX-2 has been observed in the setting of late PC induced not only by ischemia but also by delta-opioid agonists and physical exercise, supporting the view that the recruitment of this protein is a central mechanism whereby the heart protects itself from ischemia. The beneficial actions of COX-2 appear to be mediated by the synthesis of PGE(2) and/or PGI(2). Since inhibition of iNOS in preconditioned myocardium blocks COX-2 activity whereas inhibition of COX-2 does not affect iNOS activity, COX-2 appears to be downstream of iNOS in the protective pathway of late PC. The results of these studies challenge the widely accepted paradigm that views COX-2 activity as detrimental. The discovery that COX-2 plays an indispensable role in the anti-stunning and anti-infarct effects of late PC demonstrates that the recruitment of this protein is a fundamental mechanism whereby the heart adapts to stress, thereby revealing a novel, hitherto unappreciated cardioprotective function of COX-2. From a practical standpoint, the recognition that COX-2 is an obligatory co-mediator (together with iNOS) of the protection afforded by late PC has implications for the clinical use of COX-2 selective inhibitors as well as nonselective COX inhibitors. For example, the possibility that inhibition of COX-2 activity may augment myocardial cell death by obliterating the innate defensive response of the heart against ischemia/reperfusion injury needs to be considered and is the object of much current debate. Furthermore, the concept that the COX-2 byproducts, PGE(2) and/or PGI(2), play a necessary role in late PC provides a basis for novel therapeutic strategies designed to enhance the biosynthesis of these cytoprotective prostanoids in the ischemic myocardium. From a conceptual standpoint, the COX-2 hypothesis of late PC expands our understanding of the function of this enzyme in the cardiovascular system and impels a critical reassessment of current thinking regarding the biologic significance of COX-2.
Figures
Similar articles
-
Cardioprotective function of inducible nitric oxide synthase and role of nitric oxide in myocardial ischemia and preconditioning: an overview of a decade of research.J Mol Cell Cardiol. 2001 Nov;33(11):1897-918. doi: 10.1006/jmcc.2001.1462. J Mol Cell Cardiol. 2001. PMID: 11708836 Review.
-
Inducible nitric oxide synthase modulates cyclooxygenase-2 activity in the heart of conscious rabbits during the late phase of ischemic preconditioning.Circ Res. 2002 Mar 22;90(5):602-8. doi: 10.1161/01.res.0000012202.52809.40. Circ Res. 2002. PMID: 11909825
-
Cyclooxygenase-2 mediates the cardioprotective effects of the late phase of ischemic preconditioning in conscious rabbits.Proc Natl Acad Sci U S A. 2000 Aug 29;97(18):10197-202. doi: 10.1073/pnas.97.18.10197. Proc Natl Acad Sci U S A. 2000. PMID: 10963682 Free PMC article.
-
IL-6 plays an obligatory role in late preconditioning via JAK-STAT signaling and upregulation of iNOS and COX-2.Cardiovasc Res. 2004 Oct 1;64(1):61-71. doi: 10.1016/j.cardiores.2004.05.011. Cardiovasc Res. 2004. PMID: 15364614 Free PMC article.
-
Delayed adaptation of the heart to stress: late preconditioning.Stroke. 2004 Nov;35(11 Suppl 1):2676-9. doi: 10.1161/01.STR.0000143220.21382.fd. Epub 2004 Sep 30. Stroke. 2004. PMID: 15459441 Free PMC article. Review.
Cited by
-
Daily ischemic preconditioning provides sustained protection from ischemia-reperfusion induced endothelial dysfunction: a human study.J Am Heart Assoc. 2013 Feb 22;2(1):e000075. doi: 10.1161/JAHA.112.000075. J Am Heart Assoc. 2013. PMID: 23525419 Free PMC article. Clinical Trial.
-
MicroRNAs in ischemia-reperfusion injury.Am J Cardiovasc Dis. 2012;2(3):237-47. Epub 2012 Jul 25. Am J Cardiovasc Dis. 2012. PMID: 22937493 Free PMC article.
-
Anti-inflammatory therapies in myocardial infarction: failures, hopes and challenges.Br J Pharmacol. 2018 May;175(9):1377-1400. doi: 10.1111/bph.14155. Epub 2018 Mar 4. Br J Pharmacol. 2018. PMID: 29394499 Free PMC article. Review.
-
Upregulation of iNOS Protects Cyclic Mechanical Stretch-Induced Cell Death in Rat Aorta Smooth Muscle Cells.Int J Mol Sci. 2020 Nov 17;21(22):8660. doi: 10.3390/ijms21228660. Int J Mol Sci. 2020. PMID: 33212839 Free PMC article.
-
Adiponectin as an anti-inflammatory factor.Clin Chim Acta. 2007 May 1;380(1-2):24-30. doi: 10.1016/j.cca.2007.01.026. Epub 2007 Feb 2. Clin Chim Acta. 2007. PMID: 17343838 Free PMC article. Review.
References
-
- Cohen MV, Baines CP, Downey JM. Ischemic preconditioning: From adenosine receptor to KATP channel. Annu Rev Physiol. 2000;62:79–109. - PubMed
-
- Bolli R. The late phase of preconditioning. Circ Res. 2000;87:972–983. - PubMed
-
- Bolli R, Manchikalapudi S, Tang XL, et al. The protective effect of late preconditioning against myocardial stunning in conscious rabbits is mediated by nitric oxide synthase. Evidence that nitric oxide acts both as a trigger and as a mediator of the late phase of ischemic preconditioning. Circ Res. 1997;81:1094–1107. - PubMed
-
- Takano H, Manchikalapudi S, Tang XL, et al. Nitric oxide synthase is the mediator of late preconditioning against myocardial infarction in conscious rabbits. Circulation. 1998;98:441–449. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
