

Clustering patterns of cytotoxic T-lymphocyte epitopes in human immunodeficiency virus type 1 (HIV-1) proteins reveal imprints of immune evasion on HIV-1 global variation
- PMID: 12163596
- PMCID: PMC136996
- DOI: 10.1128/jvi.76.17.8757-8768.2002
Clustering patterns of cytotoxic T-lymphocyte epitopes in human immunodeficiency virus type 1 (HIV-1) proteins reveal imprints of immune evasion on HIV-1 global variation
Abstract
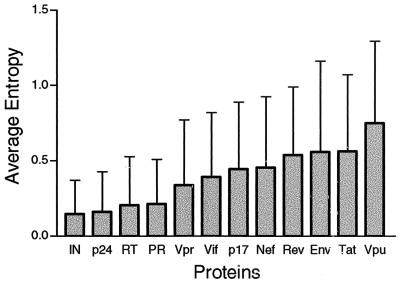
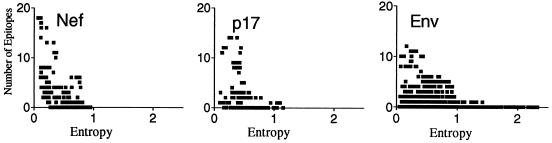
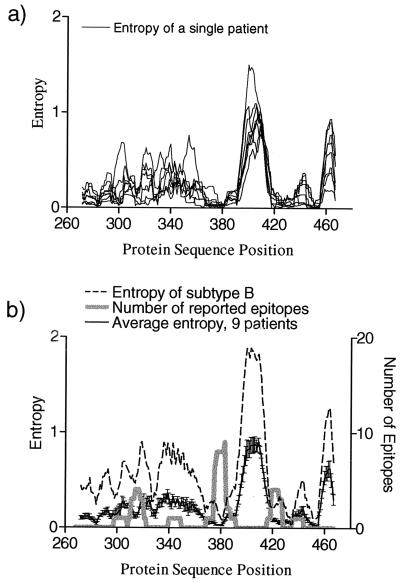
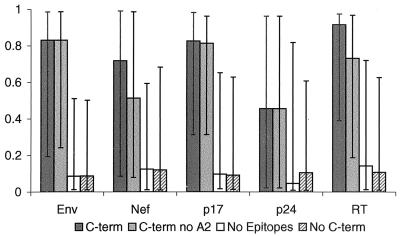
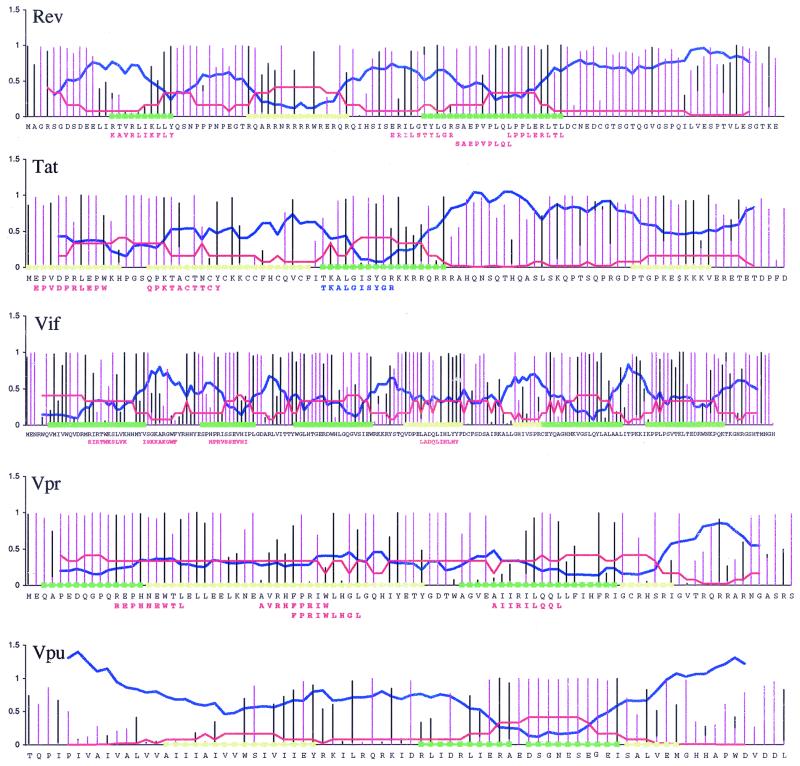
The human cytotoxic T-lymphocyte (CTL) response to human immunodeficiency virus type 1 (HIV-1) has been intensely studied, and hundreds of CTL epitopes have been experimentally defined, published, and compiled in the HIV Molecular Immunology Database. Maps of CTL epitopes on HIV-1 protein sequences reveal that defined epitopes tend to cluster. Here we integrate the global sequence and immunology databases to systematically explore the relationship between HIV-1 amino acid sequences and CTL epitope distributions. CTL responses to five HIV-1 proteins, Gag p17, Gag p24, reverse transcriptase (RT), Env, and Nef, have been particularly well characterized in the literature to date. Through comparing CTL epitope distributions in these five proteins to global protein sequence alignments, we identified distinct characteristics of HIV amino acid sequences that correlate with CTL epitope localization. First, experimentally defined HIV CTL epitopes are concentrated in relatively conserved regions. Second, the highly variable regions that lack epitopes bear cumulative evidence of past immune escape that may make them relatively refractive to CTLs: a paucity of predicted proteasome processing sites and an enrichment for amino acids that do not serve as C-terminal anchor residues. Finally, CTL epitopes are more highly concentrated in alpha-helical regions of proteins. Based on amino acid sequence characteristics, in a blinded fashion, we predicted regions in HIV regulatory and accessory proteins that would be likely to contain CTL epitopes; these predictions were then validated by comparison to new sets of experimentally defined epitopes in HIV-1 Rev, Tat, Vif, and Vpr.
Figures

References
-
- Abele, R., and R. Tampe. 1999. Function of the transport complex TAP in cellular immune recognition. Biochim. Biophys. Acta 1461:405-419. - PubMed
-
- Addo, M. M., M. Altfeld, E. S. Rosenberg, R. L. Eldridge, M. N. Philips, K. Habeeb, A. Khatri, C. Brander, G. K. Robbins, G. P. Mazzara, P. J. Goulder, and B. D. Walker. 2001. The HIV-1 regulatory proteins Tat and Rev are frequently targeted by cytotoxic T lymphocytes derived from HIV-1-infected individuals. Proc. Natl. Acad. Sci. USA 98:1781-1786. - PMC - PubMed
-
- Allen, T. M., D. H. O'Connor, P. Jing, J. L. Dzuris, B. R. Mothe, T. U. Vogel, E. Dunphy, M. E. Liebl, C. Emerson, N. Wilson, K. J. Kunstman, X. Wang, D. B. Allison, A. L. Hughes, R. C. Desrosiers, J. D. Altman, S. M. Wolinsky, A. Sette, and D. I. Watkins. 2000. Tat-specific cytotoxic T lymphocytes select for SIV escape variants during resolution of primary viraemia. Nature 407:386-390. - PubMed
-
- Altfeld, M., M. M. Addo, R. L. Eldridge, X. G. Yu, S. Thomas, A. Khatri, D. Strick, M. N. Phillips, G. B. Cohen, S. A. Islam, S. A. Kalams, C. Brander, P. J. Goulder, E. S. Rosenberg, and B. D. Walker. 2001. Vpr is preferentially targeted by CTL during HIV-1 infection. J. Immunol. 167:2743-2752. - PubMed
-
- Arold, S., P. Franken, M. P. Strub, F. Hoh, S. Benichou, R. Benarous, and C. Dumas. 1997. The crystal structure of HIV-1 Nef protein bound to the Fyn kinase SH3 domain suggests a role for this complex in altered T cell receptor signaling. Structure 5:1361-1372. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
