

The ensemble folding kinetics of protein G from an all-atom Monte Carlo simulation
- PMID: 12165568
- PMCID: PMC123229
- DOI: 10.1073/pnas.162268099
The ensemble folding kinetics of protein G from an all-atom Monte Carlo simulation
Abstract
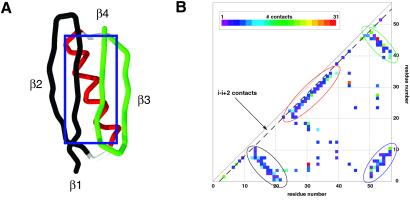
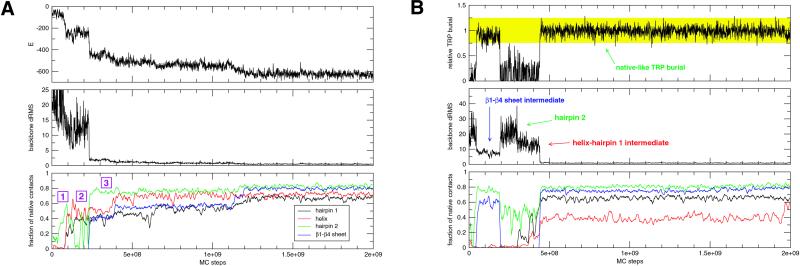
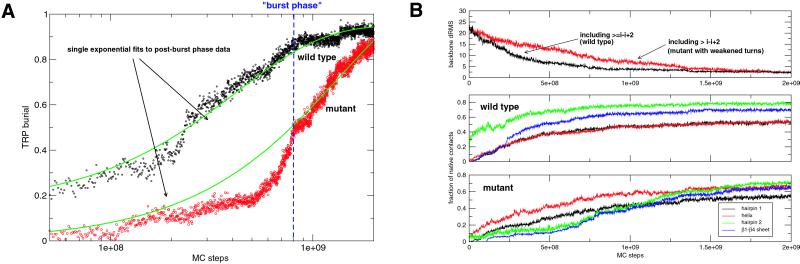
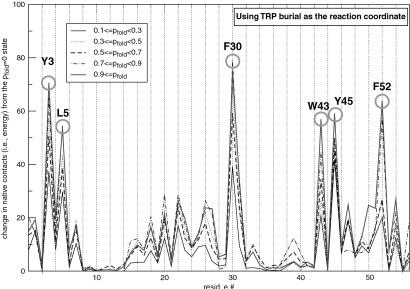
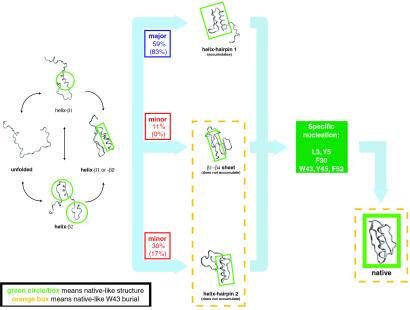
Protein G is folded with an all-atom Monte Carlo simulation by using a Gō potential. When folding is monitored by using burial of the lone tryptophan in protein G as the reaction coordinate, the ensemble kinetics is single exponential. Other experimental observations, such as the burst phase and mutational data, are also reproduced. However, more detailed analysis reveals that folding occurs over three distinct, three-state pathways. We show that, because of this tryptophan's asymmetric location in the tertiary fold, its burial (i) does not detect certain intermediates and (ii) may not correspond to the folding event. This finding demonstrates that ensemble averaging can disguise the presence of multiple pathways and intermediates when a non-ideal reaction coordinate is used. Finally, all observed folding pathways eventually converge to a common rate-limiting step, which is the formation of a specific nucleus involving hydrophobic core residues. These residues are conserved in the ubiquitin superfamily and in a phage display experiment, suggesting that fold topology is a strong determinant of the transition state.
Figures


References
-
- Duan Y. & Kollman, P. (1998) Science 282, 740-743. - PubMed
-
- Jackson S. E. (1998) Folding Des. 3, R81-R91. - PubMed
-
- Sheinerman F. B. & Brooks, C. L., III (1998) J. Mol. Biol. 278, 439-455. - PubMed
-
- Fersht A. R. & Daggett, V. (2002) Cell 108, 573-582. - PubMed
-
- Shirts M. R & Pande, V. S. (2001) Phys. Rev. Lett. 86, 4983-4987. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
