

Genome dynamics and evolution of the Mla (powdery mildew) resistance locus in barley
- PMID: 12172030
- PMCID: PMC151473
- DOI: 10.1105/tpc.002238
Genome dynamics and evolution of the Mla (powdery mildew) resistance locus in barley
Abstract
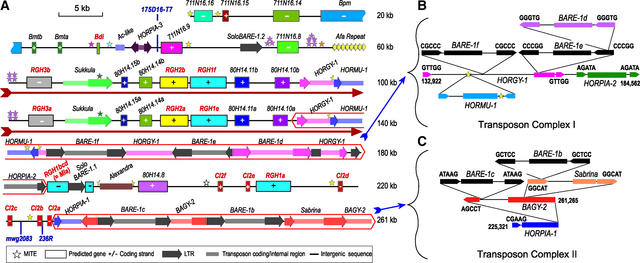
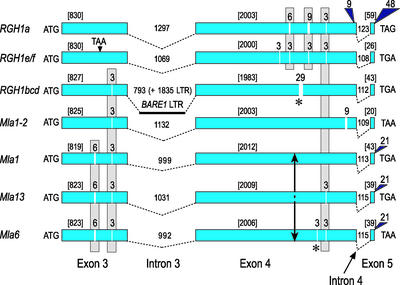
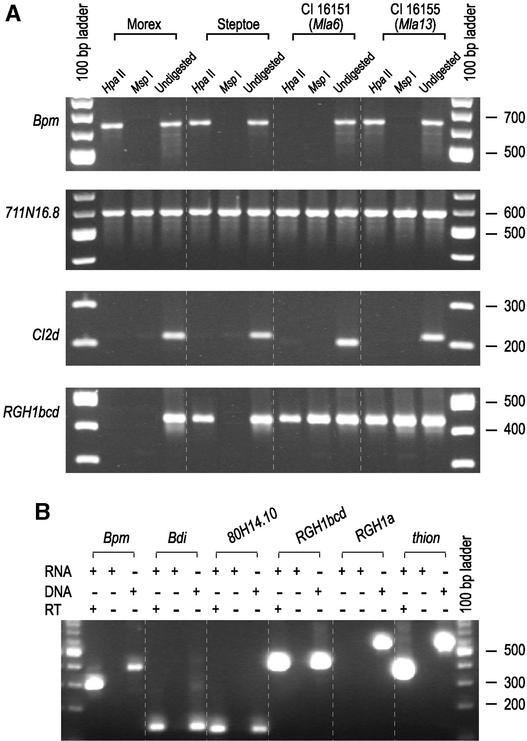
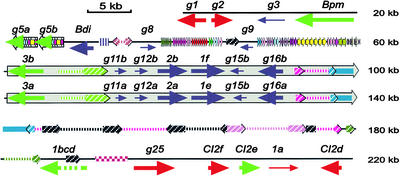
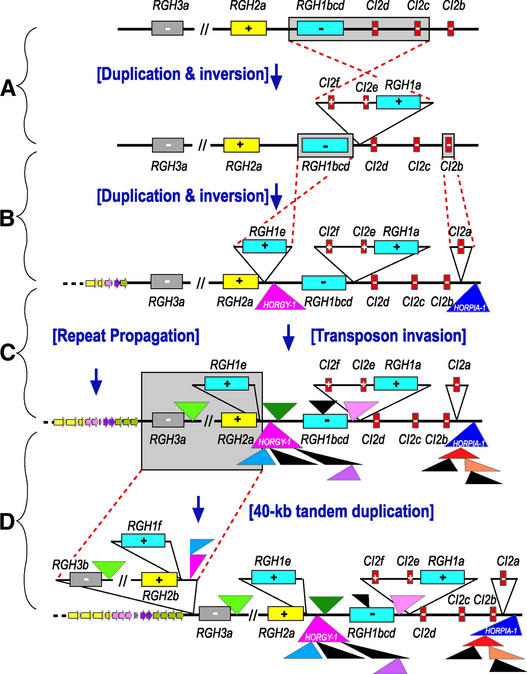
Genes that confer defense against pathogens often are clustered in the genome and evolve via diverse mechanisms. To evaluate the organization and content of a major defense gene complex in cereals, we determined the complete sequence of a 261-kb BAC contig from barley cv Morex that spans the Mla (powdery mildew) resistance locus. Among the 32 predicted genes on this contig, 15 are associated with plant defense responses; 6 of these are associated with defense responses to powdery mildew disease but function in different signaling pathways. The Mla region is organized as three gene-rich islands separated by two nested complexes of transposable elements and a 45-kb gene-poor region. A heterochromatic-like region is positioned directly proximal to Mla and is composed of a gene-poor core with 17 families of diverse tandem repeats that overlap a hypermethylated, but transcriptionally active, gene-dense island. Paleontology analysis of long terminal repeat retrotransposons indicates that the present Mla region evolved over a period of >7 million years through a variety of duplication, inversion, and transposon-insertion events. Sequence-based recombination estimates indicate that R genes positioned adjacent to nested long terminal repeat retrotransposons, such as Mla, do not favor recombination as a means of diversification. We present a model for the evolution of the Mla region that encompasses several emerging features of large cereal genomes.
Figures

References
-
- Awadalla, P., and Ritland, K. (1997). Microsatellite variation and evolution in the Mimulus guttatus species complex with contrasting mating systems. Mol. Biol. Evol. 14, 1023–1034. - PubMed
-
- Azevedo, C., Sadanandom, A., Kitagawa, K., Freialdenhoven, A., Shirasu, K., and Schulze-Lefert, P. (2002). The RAR1 interactor SGT1, an essential component of R gene-triggered disease resistance. Science 295, 2073–2076. - PubMed
-
- Baker, B., Zambriski, P., Staskawicz, B., and Dinesh-Kumar, S.P. (1997). Signaling in plant-microbe interactions. Science 276, 726–733. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
