

An in vitro model of Parkinson's disease: linking mitochondrial impairment to altered alpha-synuclein metabolism and oxidative damage
- PMID: 12177198
- PMCID: PMC6757862
- DOI: 10.1523/JNEUROSCI.22-16-07006.2002
An in vitro model of Parkinson's disease: linking mitochondrial impairment to altered alpha-synuclein metabolism and oxidative damage
Abstract
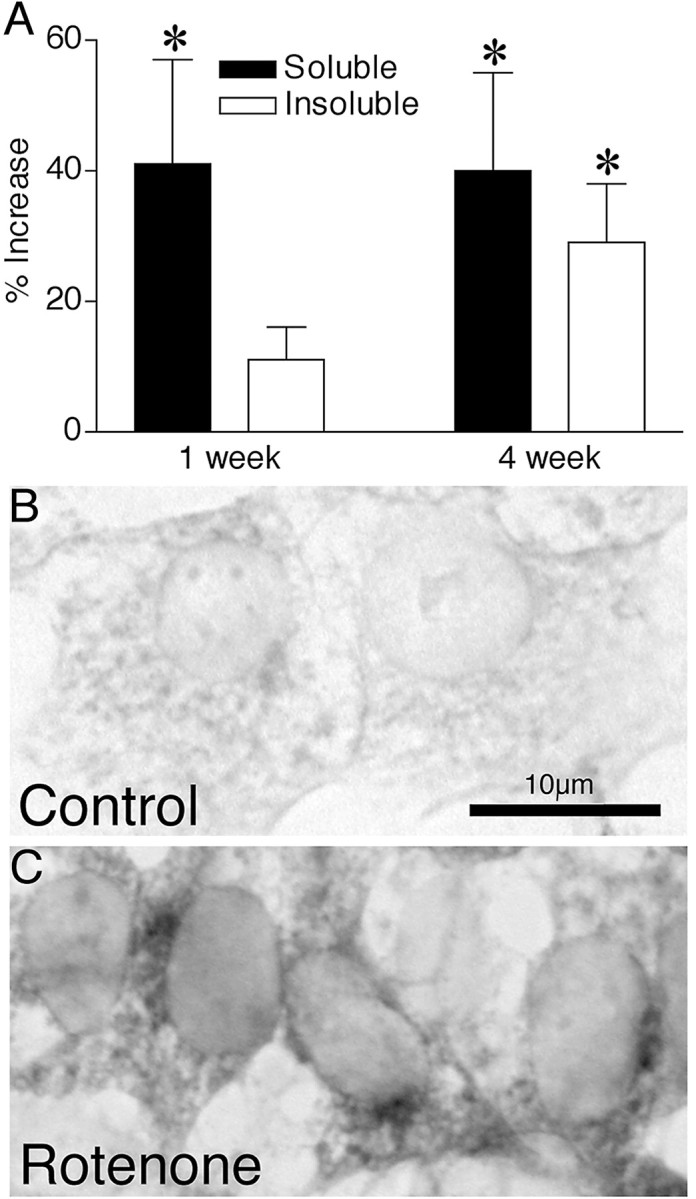
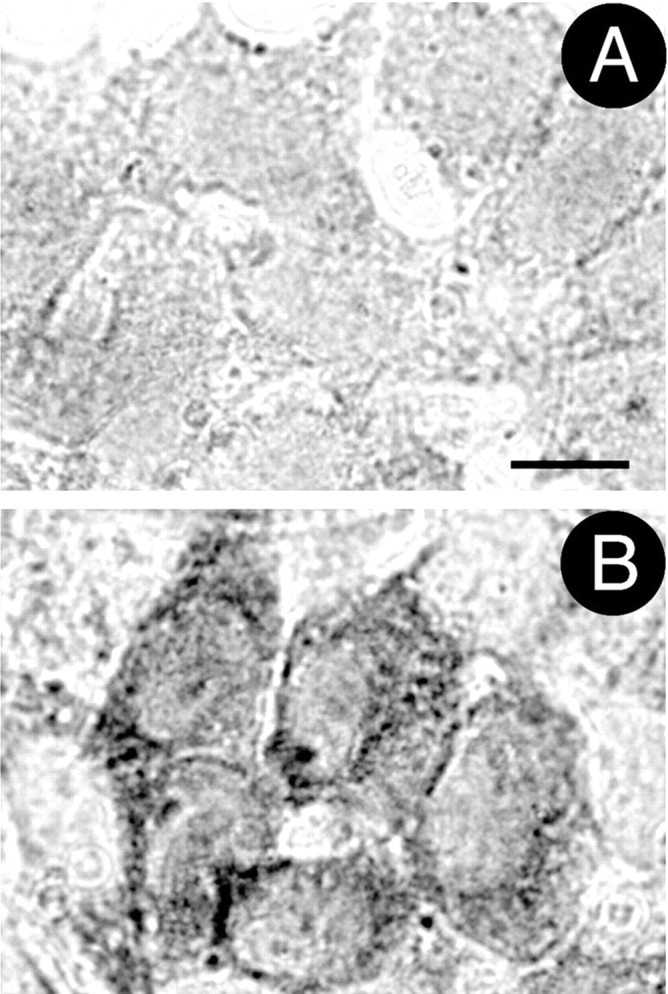
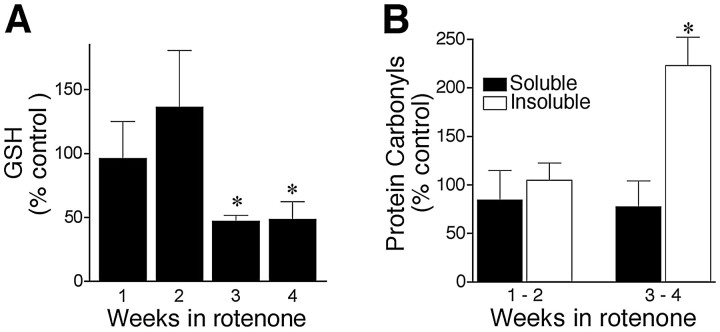
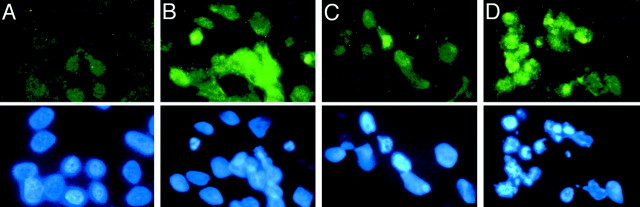
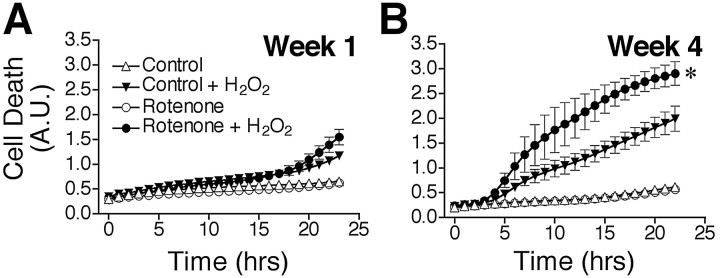
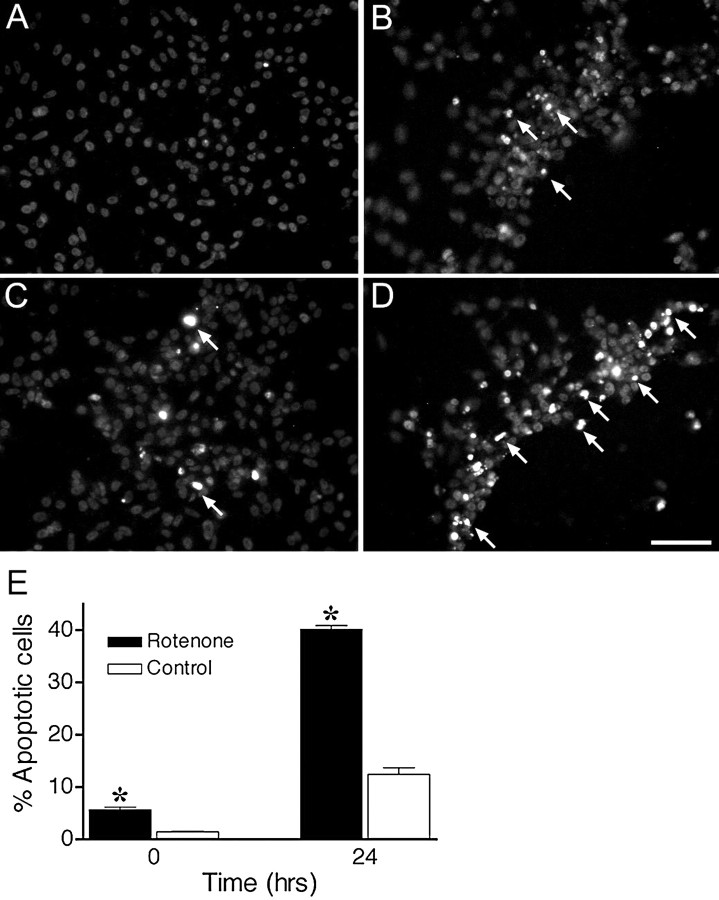
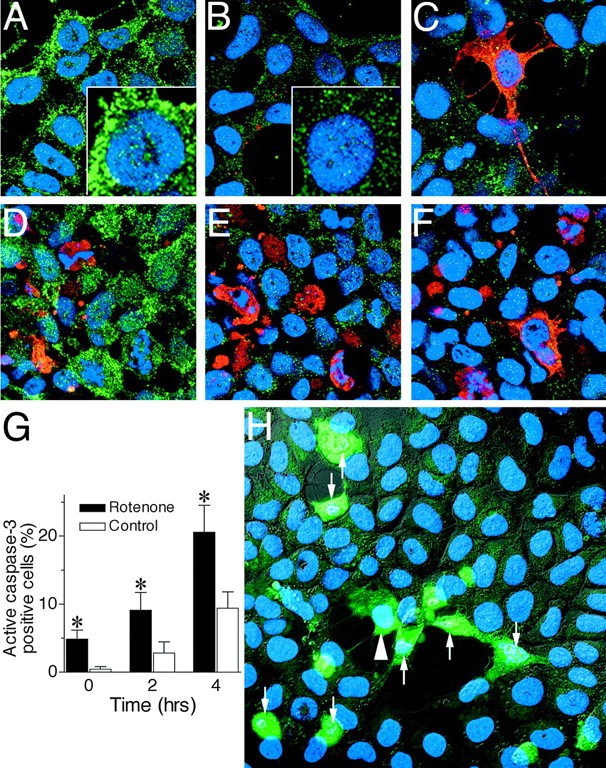
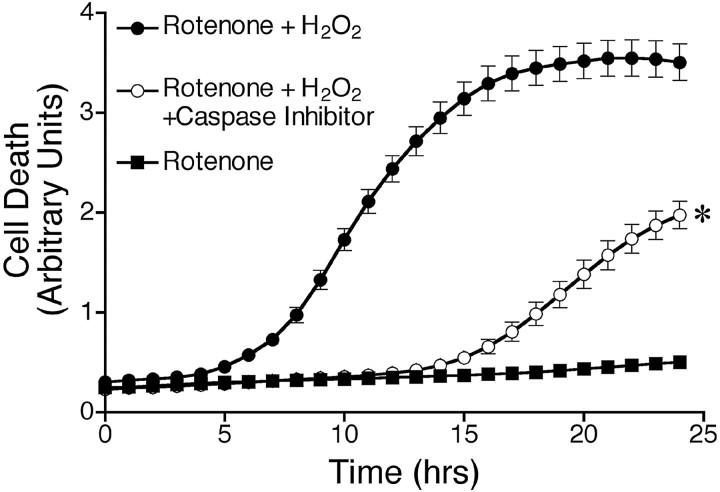
Chronic systemic complex I inhibition caused by rotenone exposure induces features of Parkinson's disease (PD) in rats, including selective nigrostriatal dopaminergic degeneration and formation of ubiquitin- and alpha-synuclein-positive inclusions (Betarbet et al., 2000). To determine underlying mechanisms of rotenone-induced cell death, we developed a chronic in vitro model based on treating human neuroblastoma cells with 5 nm rotenone for 1-4 weeks. For up to 4 weeks, cells grown in the presence of rotenone had normal morphology and growth kinetics, but at this time point, approximately 5% of cells began to undergo apoptosis. Short-term rotenone treatment (1 week) elevated soluble alpha-synuclein protein levels without changing message levels, suggesting that alpha-synuclein degradation was retarded. Chronic rotenone exposure (4 weeks) increased levels of SDS-insoluble alpha-synuclein and ubiquitin. After a latency of >2 weeks, rotenone-treated cells showed evidence of oxidative stress, including loss of glutathione and increased oxidative DNA and protein damage. Chronic rotenone treatment (4 weeks) caused a slight elevation in basal apoptosis and markedly sensitized cells to further oxidative challenge. In response to H2O2, there was cytochrome c release from mitochondria, caspase-3 activation, and apoptosis, all of which occurred earlier and to a much greater extent in rotenone-treated cells; caspase inhibition provided substantial protection. These studies indicate that chronic low-grade complex I inhibition caused by rotenone exposure induces accumulation and aggregation of alpha-synuclein and ubiquitin, progressive oxidative damage, and caspase-dependent death, mechanisms that may be central to PD pathogenesis.
Figures
Similar articles
-
Intersecting pathways to neurodegeneration in Parkinson's disease: effects of the pesticide rotenone on DJ-1, alpha-synuclein, and the ubiquitin-proteasome system.Neurobiol Dis. 2006 May;22(2):404-20. doi: 10.1016/j.nbd.2005.12.003. Epub 2006 Jan 24. Neurobiol Dis. 2006. PMID: 16439141
-
Metallothionein attenuates 3-morpholinosydnonimine (SIN-1)-induced oxidative stress in dopaminergic neurons.Antioxid Redox Signal. 2003 Jun;5(3):251-64. doi: 10.1089/152308603322110832. Antioxid Redox Signal. 2003. PMID: 12880480
-
Mechanism of toxicity in rotenone models of Parkinson's disease.J Neurosci. 2003 Nov 26;23(34):10756-64. doi: 10.1523/JNEUROSCI.23-34-10756.2003. J Neurosci. 2003. PMID: 14645467 Free PMC article.
-
Reprint of: revisiting oxidative stress and mitochondrial dysfunction in the pathogenesis of Parkinson disease-resemblance to the effect of amphetamine drugs of abuse.Free Radic Biol Med. 2013 Sep;62:186-201. doi: 10.1016/j.freeradbiomed.2013.05.042. Epub 2013 Jun 3. Free Radic Biol Med. 2013. PMID: 23743292 Review.
-
Molecular pathways of neurodegeneration in Parkinson's disease.Science. 2003 Oct 31;302(5646):819-22. doi: 10.1126/science.1087753. Science. 2003. PMID: 14593166 Review.
Cited by
-
Neuroprotective effect of nerolidol against neuroinflammation and oxidative stress induced by rotenone.BMC Neurosci. 2016 Aug 22;17(1):58. doi: 10.1186/s12868-016-0293-4. BMC Neurosci. 2016. PMID: 27549180 Free PMC article.
-
Effects of Oxidative Stress and Testosterone on Pro-Inflammatory Signaling in a Female Rat Dopaminergic Neuronal Cell Line.Endocrinology. 2016 Jul;157(7):2824-35. doi: 10.1210/en.2015-1738. Epub 2016 May 11. Endocrinology. 2016. PMID: 27167771 Free PMC article.
-
Effects of bromelain on motor responses following intra-medial forebrain bundle 6-OHDA injection in rat model of parkinsonism.Metab Brain Dis. 2019 Dec;34(6):1557-1564. doi: 10.1007/s11011-019-00462-9. Epub 2019 Jul 22. Metab Brain Dis. 2019. PMID: 31332728
-
Changes of lysosome by L-serine in rotenone-treated hippocampal neurons.Appl Microsc. 2023 Jan 10;53(1):1. doi: 10.1186/s42649-022-00084-z. Appl Microsc. 2023. PMID: 36626017 Free PMC article.
-
L-DOPA treatment from the viewpoint of neuroprotection. Possible mechanism of specific and progressive dopaminergic neuronal death in Parkinson's disease.J Neurol. 2005 Oct;252 Suppl 4:IV23-IV31. doi: 10.1007/s00415-005-4006-7. J Neurol. 2005. PMID: 16222434 Review.
References
-
- Alam ZI, Daniel SE, Lees AJ, Marsden DC, Jenner P, Halliwell B. A generalised increase in protein carbonyls in the brain in Parkinson's but not incidental Lewy body disease. J Neurochem. 1997;69:1326–1329. - PubMed
-
- Bailey SM, Pietsch EC, Cunningham CC. Ethanol stimulates the production of reactive oxygen species at mitochondrial complexes I and III. Free Radic Biol Med. 1999;27:891–900. - PubMed
-
- Barrientos A, Moraes CT. Titrating the effects of mitochondrial complex I impairment in the cell physiology. J Biol Chem. 1999;274:16188–16197. - PubMed
-
- Bennett MC, Bishop JF, Leng Y, Chock PB, Chase TN, Mouradian MM. Degradation of α-synuclein by proteasome. J Biol Chem. 1999;274:33855–33858. - PubMed
-
- Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT. Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nat Neurosci. 2000;3:1301–1306. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous