

Molecular evolution of hepatitis A virus: a new classification based on the complete VP1 protein
- PMID: 12186933
- PMCID: PMC136434
- DOI: 10.1128/jvi.76.18.9516-9525.2002
Molecular evolution of hepatitis A virus: a new classification based on the complete VP1 protein
Abstract
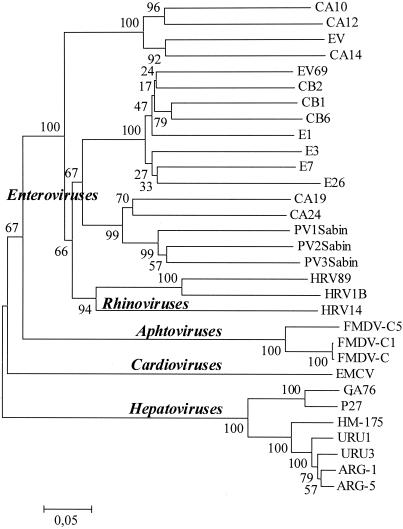
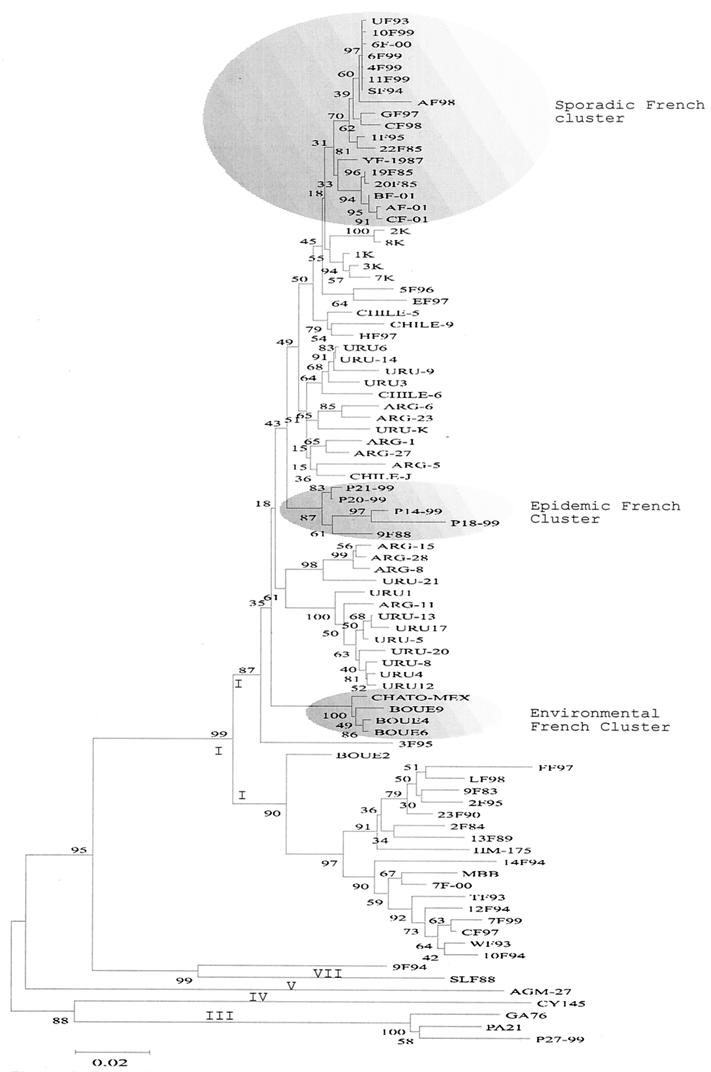
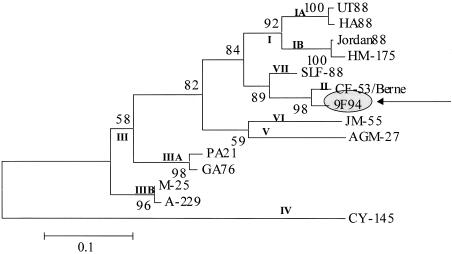
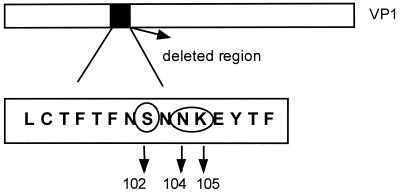
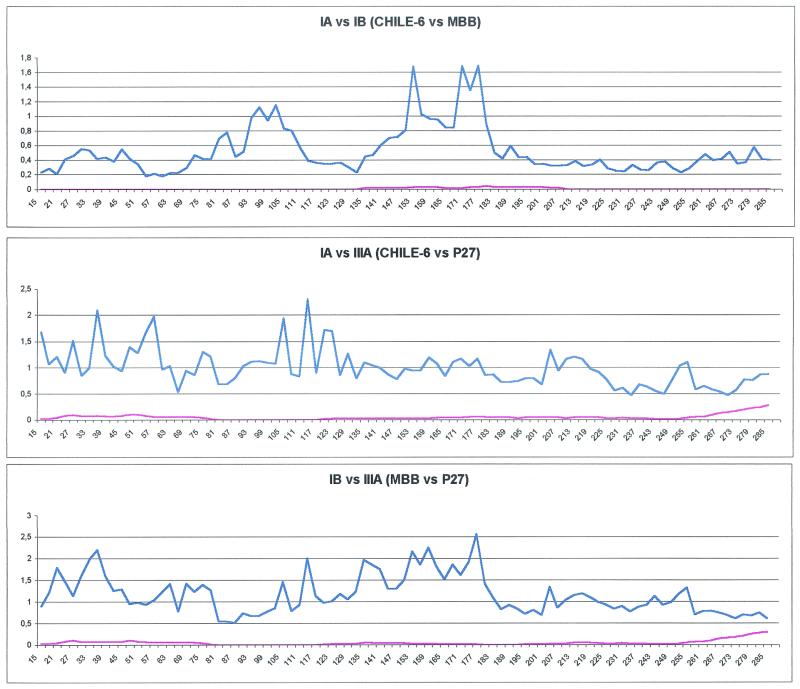
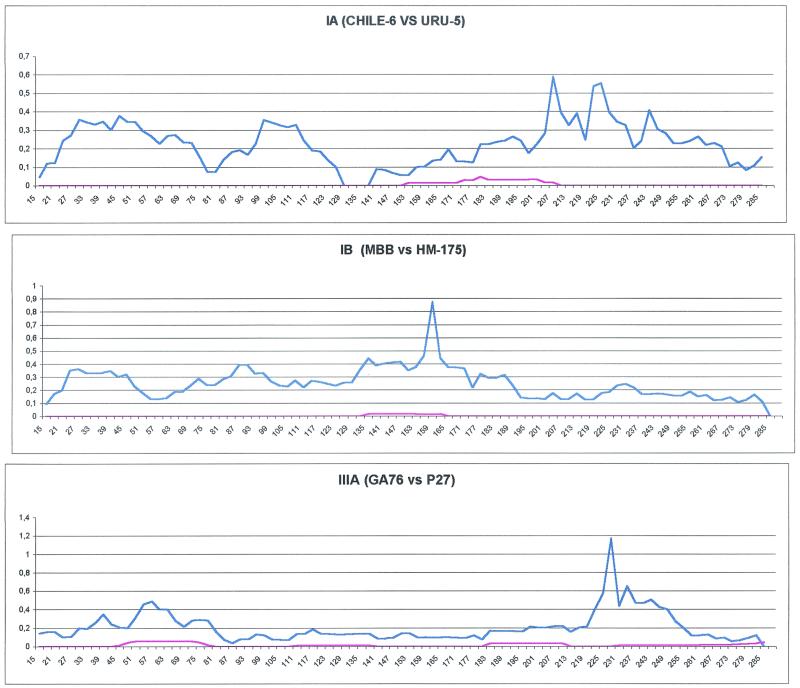
Hepatitis A virus (HAV) is a positive-stranded RNA virus in the genus Hepatovirus in the family Picornaviridae So far, analysis of the genetic variability of HAV has been based on two discrete regions, the VP1/2A junction and the VP1 N terminus. In this report, we determined the nucleotide and deduced amino acid sequences of the complete VP1 gene of 81 strains from France, Kosovo, Mexico, Argentina, Chile, and Uruguay and compared them with the sequences of seven strains of HAV isolated elsewhere. Overall strain variation in the complete VP1 gene was found to be as high as 23.7% at the nucleotide level and 10.5% at the amino acid level. Different phylogenetic methods revealed that HAV sequences form five distinct and well-supported genetic lineages. Within these lineages, HAV sequences clustered by geographical origin only for European strains. The analysis of the complete VP1 gene allowed insight into the mode of evolution of HAV and revealed the emergence of a novel variant with a 15-amino-acid deletion located on the VP1 region where neutralization escape mutations were found. This could be the first antigenic variant of HAV so far identified.
Figures
References
-
- Arauz-Ruiz, P., L. Sundqvist, Z. Garcia, L. Taylor, K. Visona, H. Norder, and L. O. Magnius. 2001. Presumed common source outbreaks of hepatitis A in an endemic area confirmed by limited sequencing within the VP1 region. J. Med. Virol. 65:449-456. - PubMed
-
- Beneduce, F., G. Pisani, M. Divizia, A. Pana, and G. Morace. 1995. Complete nucleotide sequence of a cytopathic hepatitis A virus strain isolated in Italy. Virus Res. 36:299-309. - PubMed
-
- Bosch, A., G. Sanchez, F. Le Guyader, H. Vanaclocha, L. Haugarreau, and R. M. Pinto. 2001. Human enteric viruses in coquina clams associated with a large hepatitis A outbreak. Water Sci. Technol. 43:61-65. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Medical
