

Chk2 is a tumor suppressor that regulates apoptosis in both an ataxia telangiectasia mutated (ATM)-dependent and an ATM-independent manner
- PMID: 12192050
- PMCID: PMC135625
- DOI: 10.1128/MCB.22.18.6521-6532.2002
Chk2 is a tumor suppressor that regulates apoptosis in both an ataxia telangiectasia mutated (ATM)-dependent and an ATM-independent manner
Abstract
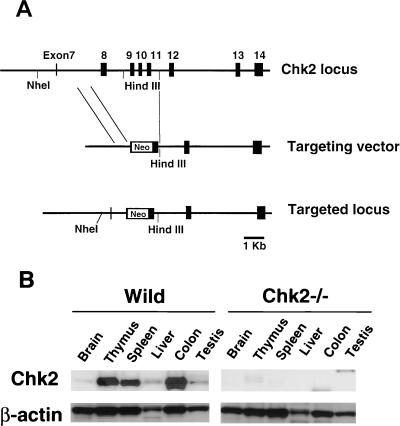
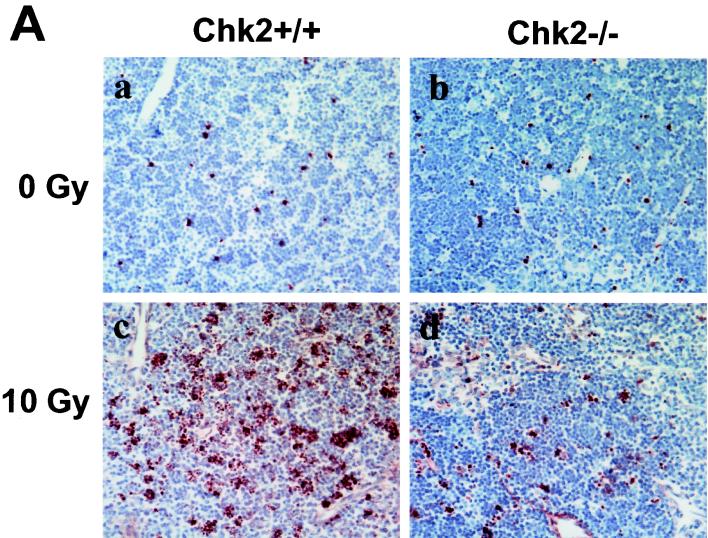
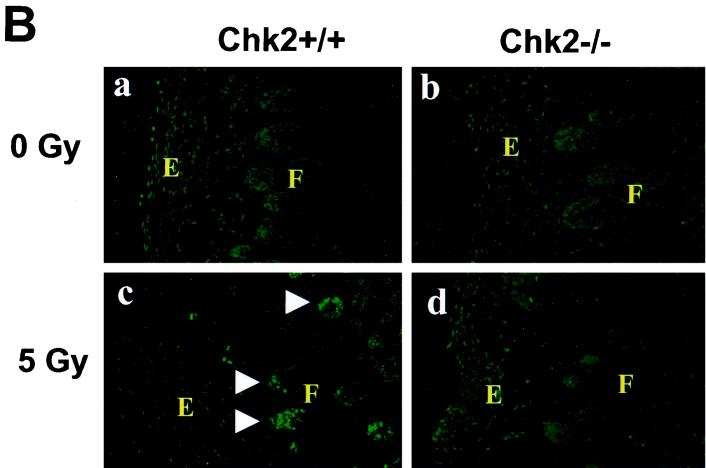
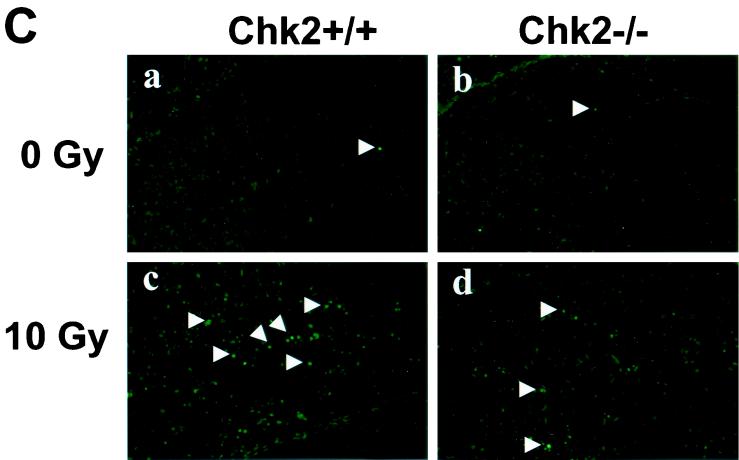
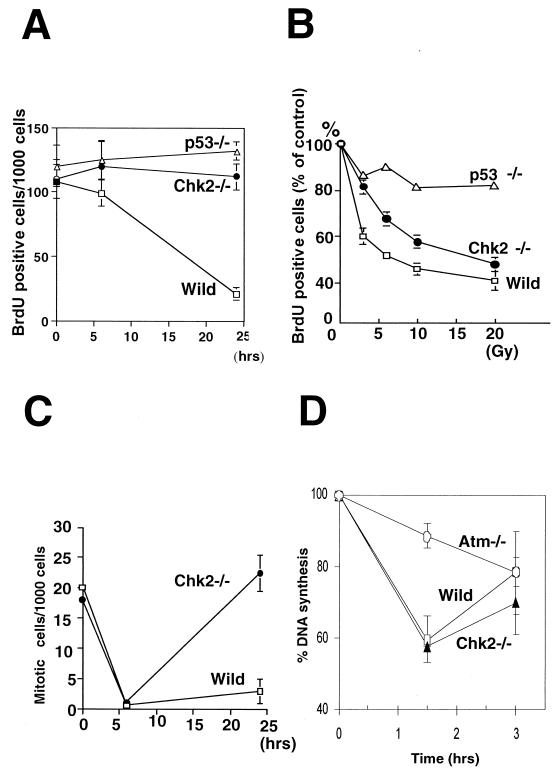
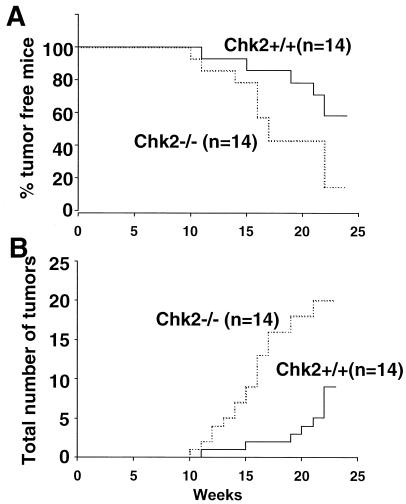
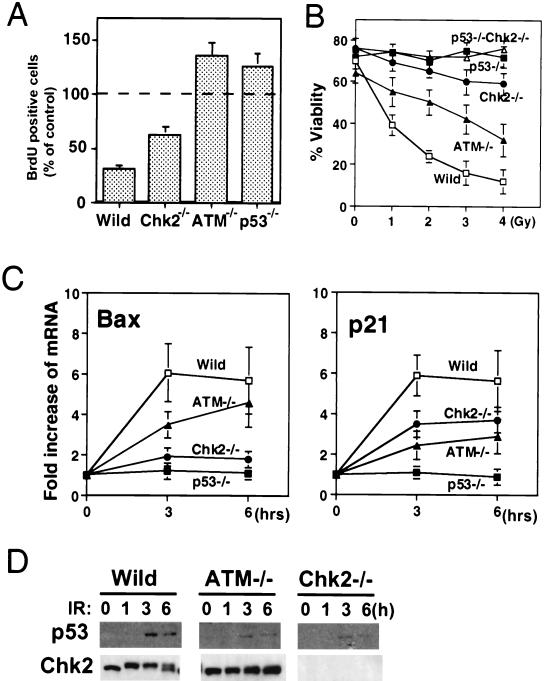
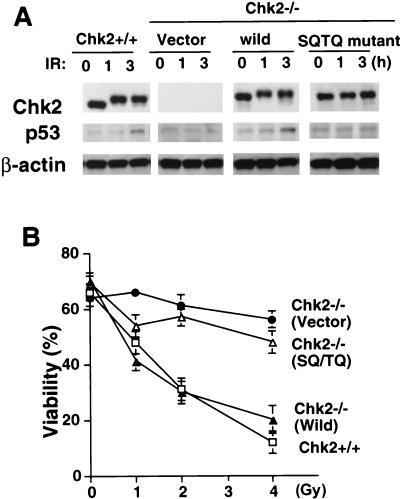
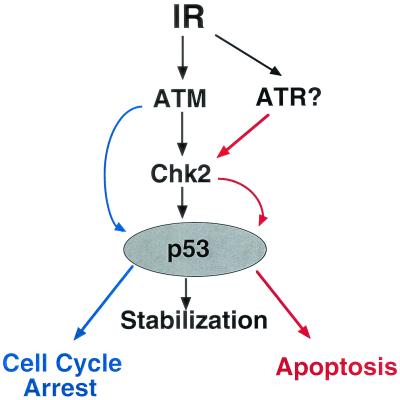
In response to ionizing radiation (IR), the tumor suppressor p53 is stabilized and promotes either cell cycle arrest or apoptosis. Chk2 activated by IR contributes to this stabilization, possibly by direct phosphorylation. Like p53, Chk2 is mutated in patients with Li-Fraumeni syndrome. Since the ataxia telangiectasia mutated (ATM) gene is required for IR-induced activation of Chk2, it has been assumed that ATM and Chk2 act in a linear pathway leading to p53 activation. To clarify the role of Chk2 in tumorigenesis, we generated gene-targeted Chk2-deficient mice. Unlike ATM(-/-) and p53(-/-) mice, Chk2(-/-) mice do not spontaneously develop tumors, although Chk2 does suppress 7,12-dimethylbenzanthracene-induced skin tumors. Tissues from Chk2(-/-) mice, including those from the thymus, central nervous system, fibroblasts, epidermis, and hair follicles, show significant defects in IR-induced apoptosis or impaired G(1)/S arrest. Quantitative comparison of the G(1)/S checkpoint, apoptosis, and expression of p53 proteins in Chk2(-/-) versus ATM(-/-) thymocytes suggested that Chk2 can regulate p53-dependent apoptosis in an ATM-independent manner. IR-induced apoptosis was restored in Chk2(-/-) thymocytes by reintroduction of the wild-type Chk2 gene but not by a Chk2 gene in which the sites phosphorylated by ATM and ataxia telangiectasia and rad3(+) related (ATR) were mutated to alanine. ATR may thus selectively contribute to p53-mediated apoptosis. These data indicate that distinct pathways regulate the activation of p53 leading to cell cycle arrest or apoptosis.
Figures
Comment in
-
CHK2: a tumor suppressor or not?Cell Cycle. 2002 Nov-Dec;1(6):401-3. doi: 10.4161/cc.1.6.264. Cell Cycle. 2002. PMID: 12548013 Review. No abstract available.
References
-
- Ahn, J. Y., J. K. Schwarz, H. Piwnica-Worms, and C. E. Canman. 2000. Threonine 68 phosphorylation by ataxia telangiectasia mutated is required for efficient activation of Chk2 in response to ionizing radiation. Cancer Res. 60:5934-5936. - PubMed
-
- Banin, S., L. Moyal, S. Shieh, Y. Taya, C. W. Anderson, L. Chessa, N. I. Smorodinsky, C. Prives, Y. Reiss, Y. Shiloh, and Y. Ziv. 1998. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science 281:1674-1677. - PubMed
-
- Barlow, C., K. D. Brown, C. X. Deng, D. A. Tagle, and A. Wynshaw-Boris. 1997. Atm selectively regulates distinct p53-dependent cell-cycle checkpoint and apoptotic pathways. Nat. Genet. 17:453-456. - PubMed
-
- Barlow, C., S. Hirotsune, R. Paylor, M. Liyanage, M. Eckhaus, F. Collins, Y. Shiloh, J. N. Crawley, T. Ried, D. Tagle, and A. Wynshaw-Boris. 1996. Atm-deficient mice: a paradigm of ataxia telangiectasia. Cell 86:159-171. - PubMed
-
- Bell, D. W., J. M. Varley, T. E. Szydlo, D. H. Kang, D. C. Wahrer, K. E. Shannon, M. Lubratovich, S. J. Verselis, K. J. Isselbacher, J. F. Fraumeni, J. M. Birch, F. P. Li, J. E. Garber, and D. A. Haber. 1999. Heterozygous germ line hCHK2 mutations in Li-Fraumeni syndrome. Science 286:2528-2531. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous