

Mutations of the selenoprotein N gene, which is implicated in rigid spine muscular dystrophy, cause the classical phenotype of multiminicore disease: reassessing the nosology of early-onset myopathies
- PMID: 12192640
- PMCID: PMC378532
- DOI: 10.1086/342719
Mutations of the selenoprotein N gene, which is implicated in rigid spine muscular dystrophy, cause the classical phenotype of multiminicore disease: reassessing the nosology of early-onset myopathies
Abstract
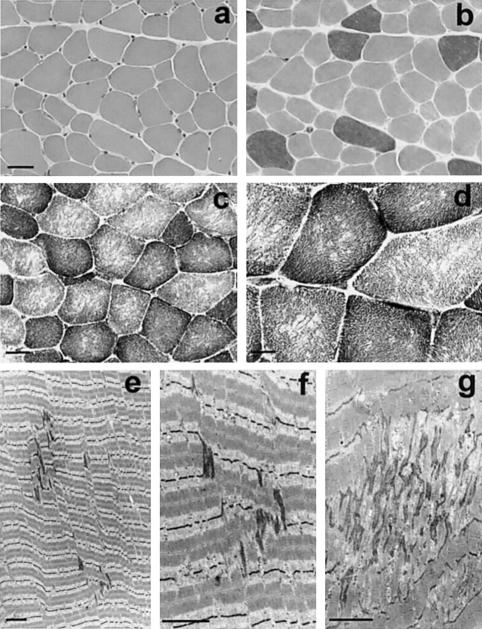
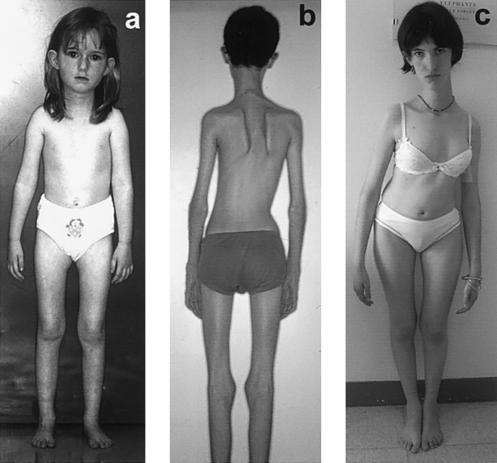
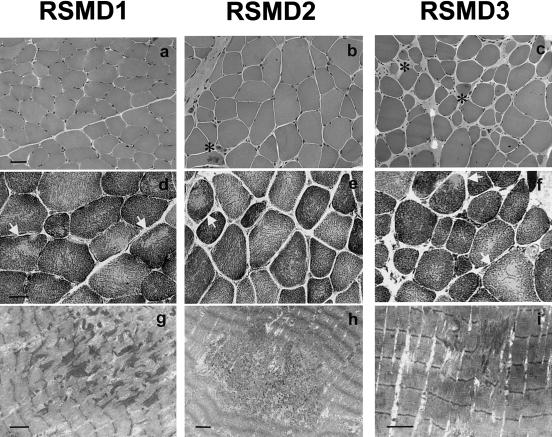
Multiminicore disease (MmD) is an autosomal recessive congenital myopathy characterized by the presence of multiple, short core lesions (known as "minicores") in most muscle fibers. MmD is a clinically heterogeneous condition, in which four subgroups have been distinguished. Homozygous RYR1 mutations have been recently identified in the moderate form of MmD with hand involvement. The genes responsible for the three other forms (including the most prevalent phenotype, termed the "classical" phenotype) remained, so far, unknown. To further characterize the genetic basis of MmD, we analyzed a series of 62 patients through a combined positional/candidate-gene approach. On the basis of clinical and morphological data, we suspected a relationship between classical MmD and the selenoprotein N gene (SEPN1), which is located on chromosome 1p36 (RSMD1 locus) and is responsible for the congenital muscular dystrophy with rigid spine syndrome (RSMD). A genomewide screening, followed by the analysis of 1p36 microsatellite markers in 27 informative families with MmD, demonstrated linkage to RSMD1 in eight families. All showed an axial myopathy with scoliosis and respiratory failure, consistent with the most severe end of the classical MmD spectrum; spinal rigidity was evident in some, but not all, patients. We excluded linkage to RSMD1 in 19 families with MmD, including 9 with classical MmD. Screening of SEPN1 in the 8 families that showed linkage and in 14 patients with classical sporadic disease disclosed 9 mutations affecting 17 patients (12 families); 6 were novel mutations, and 3 had been described in patients with RSMD. Analysis of three deltoid biopsy specimens from patients with typical RSMD revealed a wide myopathological variability, ranging from a dystrophic to a congenital myopathy pattern. A variable proportion of minicores was found in all the samples. The present study represents the first identification of a gene responsible for classical MmD, demonstrates its genetic heterogeneity, and reassesses the nosological boundaries between MmD and RSMD.
Figures
References
Electronic-Database Information
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for RSMD1 [MIM 602771], SEPN1 [MIM 606210], minicore myopathy with external ophthalmoplegia [MIM 255320], and RYR1 [MIM 180901])
-
- GenBank, http://www.ncbi.nlm.nih.gov/Genbank/ (for SEPN1 genomic sequence [accession number AJ306398] and SEPN1 cDNA sequence [accession number AJ306399])
References
-
- Banker BQ (1994) The congenital muscular dystrophies. In: Engel AG, Franzini-Amstrong C (eds) Myology. Vol 2. McGraw-Hill, New York, pp 1275–1289
-
- Behne D, Kyriakopoulos A (2001) Mammalian selenium-containing proteins. Annu Rev Nutr 21:453–473 - PubMed
-
- Ben Hamida M, Hentati F, Ben Hamida C (1987) Maladie à multiminicores au cours d'un syndrome de la colonne raide. Rev Neurol (Paris) 143:284–289 - PubMed
-
- Brown MR, Cohen HJ, Lyons JM, Curtis TW, Thunberg B, Cochran WJ, Klish WJ (1986) Proximal muscle weakness and selenium deficiency associated with long term parenteral nutrition. Am J Clin Nutr 43:549–554 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
