

Nf-kappa B, chemokine gene transcription and tumour growth
- PMID: 12209135
- PMCID: PMC2668257
- DOI: 10.1038/nri887
Nf-kappa B, chemokine gene transcription and tumour growth
Abstract
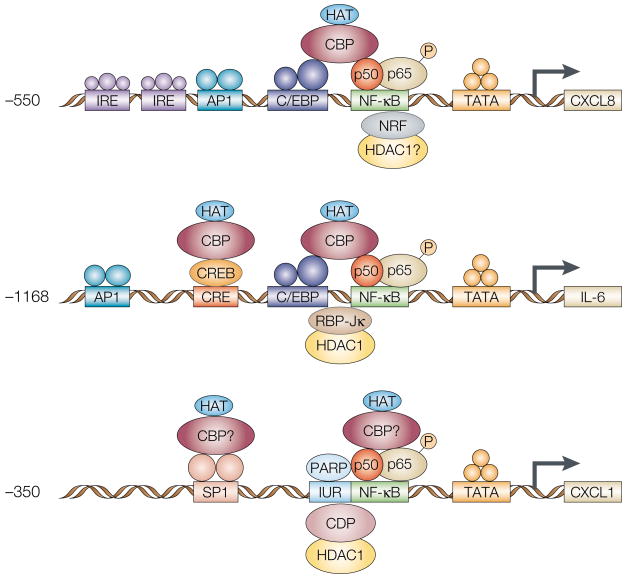
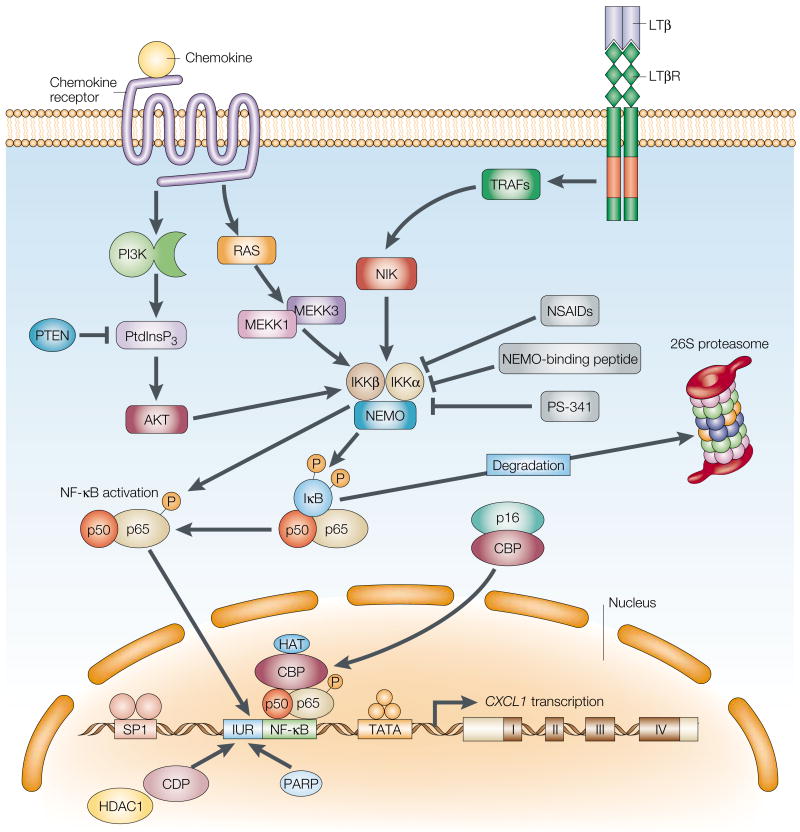
The constitutive expression of angiogenic and tumorigenic chemokines by tumour cells facilitates the growth of tumours. The transcription of these angiogenic and tumorigenic chemokine genes is modulated, in part, by the nuclear factor-kappa B (NF-kappa B) family of transcription factors. In some tumours, there is constitutive activation of the kinases that modulate the activity of inhibitor of NF-kappa B (I kappa B) kinase (IKK), which leads to the constitutive activation of members of the NF-kappa B family. This activation of NF-kappa B is associated with the dysregulation of transcription of genes that encode cytokines, chemokines, adhesion factors and inhibitors of apoptosis. In this review, I discuss the factors that lie upstream of the NF-kappa B cascade that are activated during tumorigenesis and the role of the putative NF-kappa B enhanceosome in constitutive chemokine gene transcription during tumorigenesis.
Figures

References
-
- Rossi D, Zlotnik A. The biology of chemokines and their receptors. Annu Rev Immunol. 2000;18:217–242. - PubMed
-
- Luster AD. The role of chemokines in linking innate and adaptive immunity. Curr Opin Immunol. 2002;14:129–135. - PubMed
-
- Homey B, Muller A, Zlotnik A. Chemokines: agents for the immunotherapy of cancer. Nature Rev Immunol. 2002;2:175–184. - PubMed
-
- Devalaraja MN, Richmond A. Multiple chemotactic factors, fine control or redundancy? Trends Pharmacol Sci. 1999;20:151–156. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
