

Protein kinase C-alpha and ERK1/2 mediate mitochondrial dysfunction, decreases in active Na+ transport, and cisplatin-induced apoptosis in renal cells
- PMID: 12218054
- PMCID: PMC1948818
- DOI: 10.1074/jbc.M206373200
Protein kinase C-alpha and ERK1/2 mediate mitochondrial dysfunction, decreases in active Na+ transport, and cisplatin-induced apoptosis in renal cells
Abstract
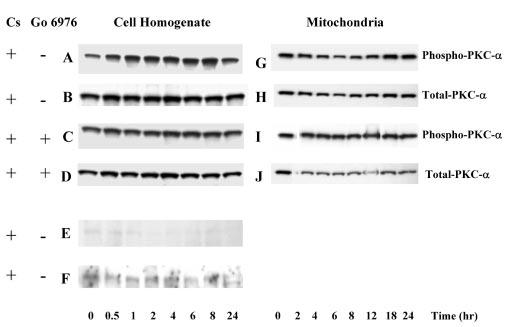
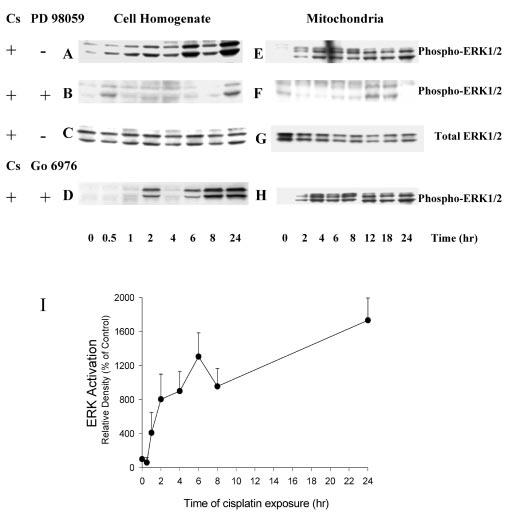
Initiation of apoptosis by many agents is preceded by mitochondrial dysfunction and depolarization of the mitochondrial inner membrane. Here we demonstrate that, in renal proximal tubular cells (RPTC), cisplatin induces mitochondrial dysfunction associated with hyperpolarization of the mitochondrial membrane and that these events are mediated by protein kinase C (PKC)-alpha and ERK1/2. Cisplatin induced sustained decreases in RPTC respiration, oxidative phosphorylation, and increases in the mitochondrial transmembrane potential (deltaPsi(m)), which were preceded by the inhibition of F(0)F(1)-ATPase and cytochrome c release from the mitochondria, accompanied by caspase-3 activation, and followed by RPTC apoptosis. Cisplatin also decreased active Na+ transport as a result, in part, of the inhibition of Na+/K(+)-ATPase. These changes were preceded by PKC-alpha and ERK1/2 activation. Inhibition of cisplatin-induced PKC-alpha and ERK1/2 activation using Go6976 and PD98059, respectively, abolished increases in deltaPsi(m), diminished decreases in oxidative phosphorylation, active Na+ transport, and decreased caspase-3 activation without blocking cytochrome c release. Caspase inhibitor benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone (zVAD-fmk) did not prevent increases in deltaPsi(m). Furthermore, inhibition of PKC-alpha did not prevent cisplatin-induced ERK1/2 activation. We concluded that in RPTC: 1) cisplatin-induced mitochondrial dysfunction, decreases in active Na+ transport, and apoptosis are mediated by PKC-alpha and ERK1/2; 2) PKC-alpha and ERK1/2 mediate activation of caspase-3 by acting downstream of cytochrome c release from mitochondria; and 3) ERK1/2 activation by cisplatin occurs through a PKC-alpha-independent pathway.
Figures











References
-
- Goldstein RS, Mayor GH. Life Sci. 1983;32:685–690. - PubMed
-
- Safirstein RL, Winston J, Moel D, Dikman S, Guttenplan J. Int. J. Androl. 1987;10:325–346. - PubMed
-
- Miura K, Goldstein RS, Pasino DA, Hook JB. Toxicology. 1987;44:147–158. - PubMed
-
- Ikeda S, Fukuzaki A, Kaneto H, Ishidoya S, Orikasa S. Int. J. Urol. 1999;6:245–250. - PubMed
-
- Osman AM, El-Sayed EM, El-Demerdash E, Al-Hyder A, El-Didi M, Attia AS, Hamada FM. Pharm. Res. 2000;41:115–121. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
