

Homologous recombination resolution defect in werner syndrome
- PMID: 12242278
- PMCID: PMC139822
- DOI: 10.1128/MCB.22.20.6971-6978.2002
Homologous recombination resolution defect in werner syndrome
Abstract
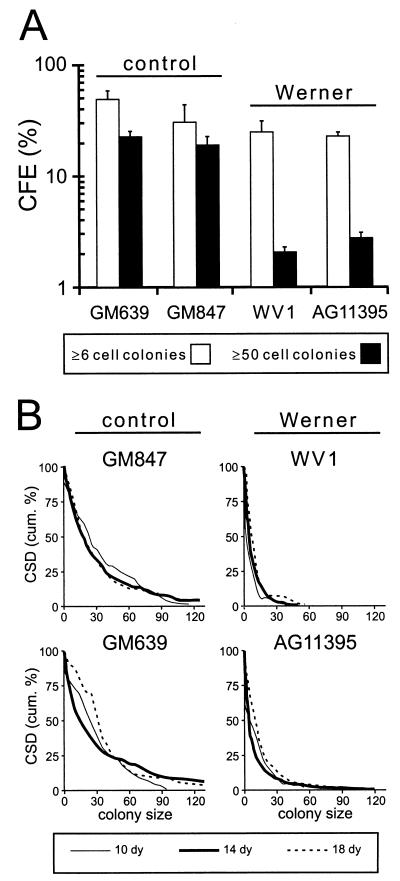
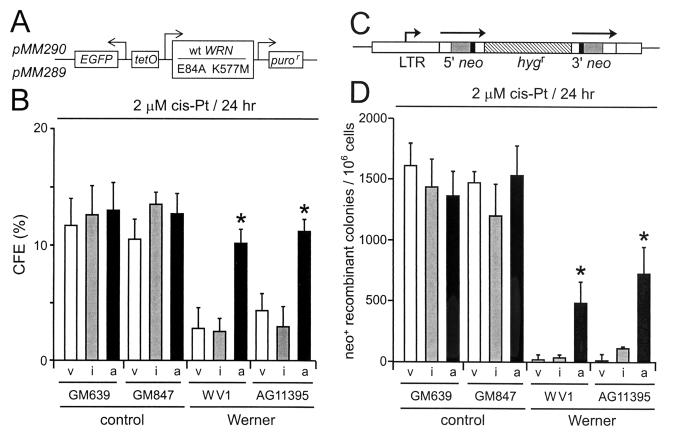
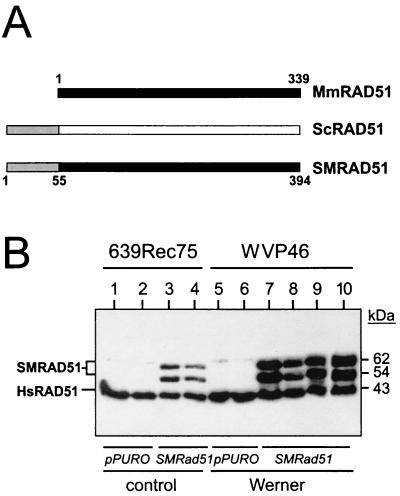
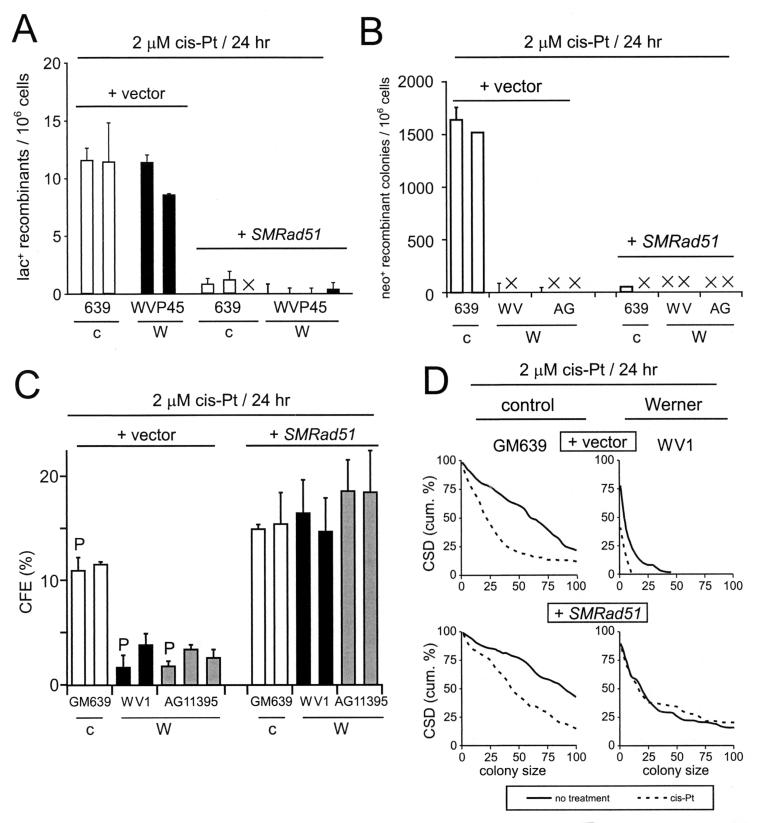
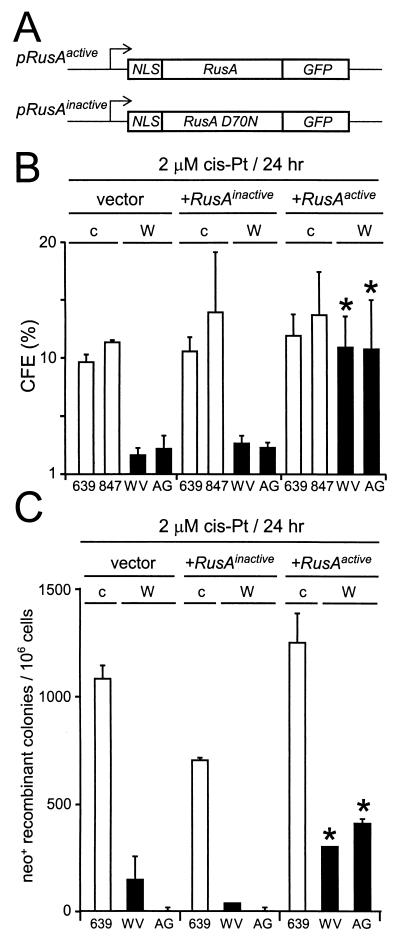
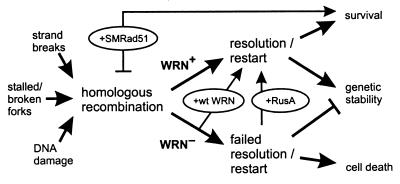
Werner syndrome (WRN) is an uncommon autosomal recessive disease whose phenotype includes features of premature aging, genetic instability, and an elevated risk of cancer. We used three different experimental strategies to show that WRN cellular phenotypes of limited cell division potential, DNA damage hypersensitivity, and defective homologous recombination (HR) are interrelated. WRN cell survival and the generation of viable mitotic recombinant progeny could be rescued by expressing wild-type WRN protein or by expressing the bacterial resolvase protein RusA. The dependence of WRN cellular phenotypes on RAD51-dependent HR pathways was demonstrated by using a dominant-negative RAD51 protein to suppress mitotic recombination in WRN and control cells: the suppression of RAD51-dependent recombination led to significantly improved survival of WRN cells following DNA damage. These results define a physiological role for the WRN RecQ helicase protein in RAD51-dependent HR and identify a mechanistic link between defective recombination resolution and limited cell division potential, DNA damage hypersensitivity, and genetic instability in human somatic cells.
Figures
References
-
- Bennett, S. E., A. Umar, J. Oshima, R. J. Monnat, Jr., and T. A. Kunkel. 1997. Mismatch repair in extracts of Werner syndrome cell lines. Cancer Res. 57:2956-2960. - PubMed
-
- Boddy, M. N., P.-H. L. Gaillard, W. H. McDonald, P. Shanahan, J. R. I. Yates, and P. Russell. 2001. Mus81-Eme1 are essential components of a Holliday junction resolvase. Cell 107:537-548. - PubMed
-
- Brooks-Wilson, A. R., M. J. Emond, and R. J. Monnat, Jr. 1997. Unexpectedly low loss of heterozygosity in genetically unstable Werner syndrome cell lines. Genes Chromosomes Cancer 18:133-142. - PubMed
-
- Brosh, R. M., Jr., and V. A. Bohr. 2002. Roles of the Werner syndrome protein in pathways required for maintenance of genome stability. Exp. Gerontol. 37:491-506. - PubMed
-
- Chen, X.-B., R. Melchionna, C.-M. Denis, P.-H. L. Gaillard, A. Blasina, I. V. de Weyer, M. N. Boddy, P. Russell, J. Vialard, and C. H. McGowan. 2001. Human Mus81-associated endonuclease cleaves Holliday junctions in vitro. Mol. Cell 8:1117-1127. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials