

Genomic rearrangements resulting in PLP1 deletion occur by nonhomologous end joining and cause different dysmyelinating phenotypes in males and females
- PMID: 12297985
- PMCID: PMC378540
- DOI: 10.1086/342728
Genomic rearrangements resulting in PLP1 deletion occur by nonhomologous end joining and cause different dysmyelinating phenotypes in males and females
Abstract
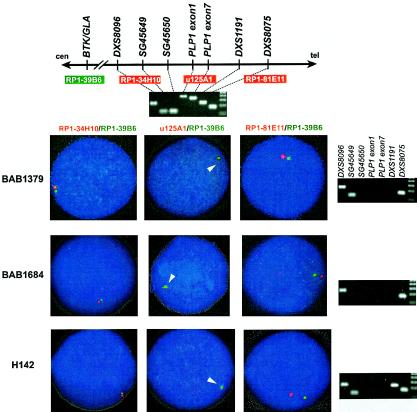
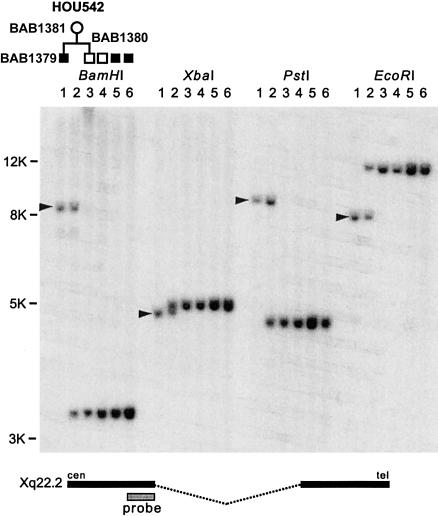
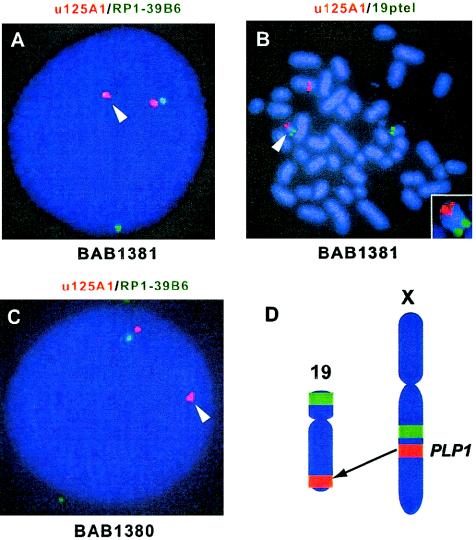
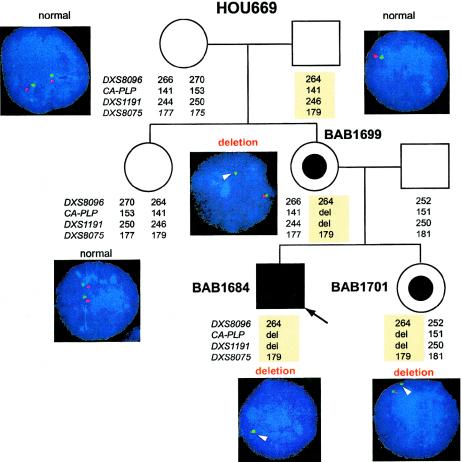
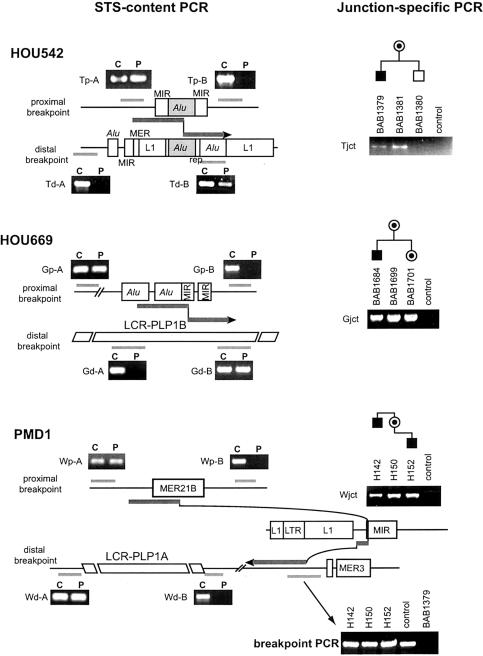
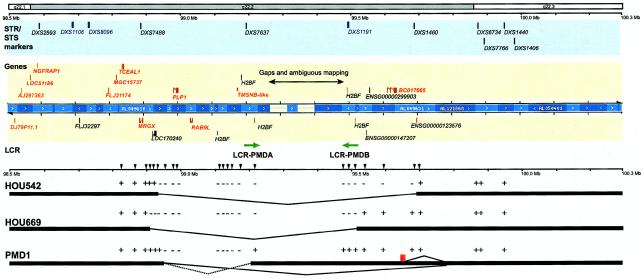
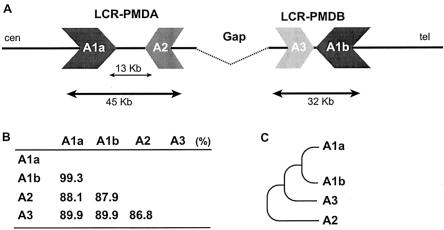
In the majority of patients with Pelizaeus-Merzbacher disease, duplication of the proteolipid protein gene PLP1 is responsible, whereas deletion of PLP1 is infrequent. Genomic mechanisms for these submicroscopic chromosomal rearrangements remain unknown. We identified three families with PLP1 deletions (including one family described elsewhere) that arose by three distinct processes. In one family, PLP1 deletion resulted from a maternal balanced submicroscopic insertional translocation of the entire PLP1 gene to the telomere of chromosome 19. PLP1 on the 19qtel is probably inactive by virtue of a position effect, because a healthy male sibling carries the same der(19) chromosome along with a normal X chromosome. Genomic mapping of the deleted segments revealed that the deletions are smaller than most of the PLP1 duplications and involve only two other genes. We hypothesize that the deletion is infrequent, because only the smaller deletions can avoid causing either infertility or lethality. Analyses of the DNA sequence flanking the deletion breakpoints revealed Alu-Alu recombination in the family with translocation. In the other two families, no homologous sequence flanking the breakpoints was found, but the distal breakpoints were embedded in novel low-copy repeats, suggesting the potential involvement of genome architecture in stimulating these rearrangements. In one family, junction sequences revealed a complex recombination event. Our data suggest that PLP1 deletions are likely caused by nonhomologous end joining.
Figures

References
Electronic-Database Information
-
- BLAST, http://www.ncbi.nlm.nih.gov/BLAST/ (for sequence identity analysis)
-
- Fgenesh, http://genomic.sanger.ac.uk/gf/gf.shtml (for sequence analysis)
-
- Grail, http://grail.lsd.ornl.gov/ (for sequence analysis)
-
- MZEF, http://argon.cshl.org/genefinder/ (for sequence analysis)
References
-
- Baumbach LL, Chamberlain JS, Ward PA, Farwell NJ, Caskey CT (1989) Molecular and clinical correlation of deletion leading to Duchenne and Becker muscular dystrophies. Neurology 39:465–474 - PubMed
-
- Baur JA, Zou Y, Shay JW, Wright WE (2001) Telomere position effect in human cells. Science 292:2075–2077 - PubMed
-
- Eichler EE (2001) Segmental duplications: what's missing, misassigned, and misassembled—and should we care? Genome Res 11:653–656 - PubMed
-
- Ellis D, Malcolm S (1994) Proteolipid protein gene dosage effect in Pelizaeus-Merzbacher disease. Nat Genet 6:333–334 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
