

Activation of Bcl-2-associated death protein and counter-response of Akt within cell populations during seizure-induced neuronal death
- PMID: 12351720
- PMCID: PMC6757765
- DOI: 10.1523/JNEUROSCI.22-19-08458.2002
Activation of Bcl-2-associated death protein and counter-response of Akt within cell populations during seizure-induced neuronal death
Abstract
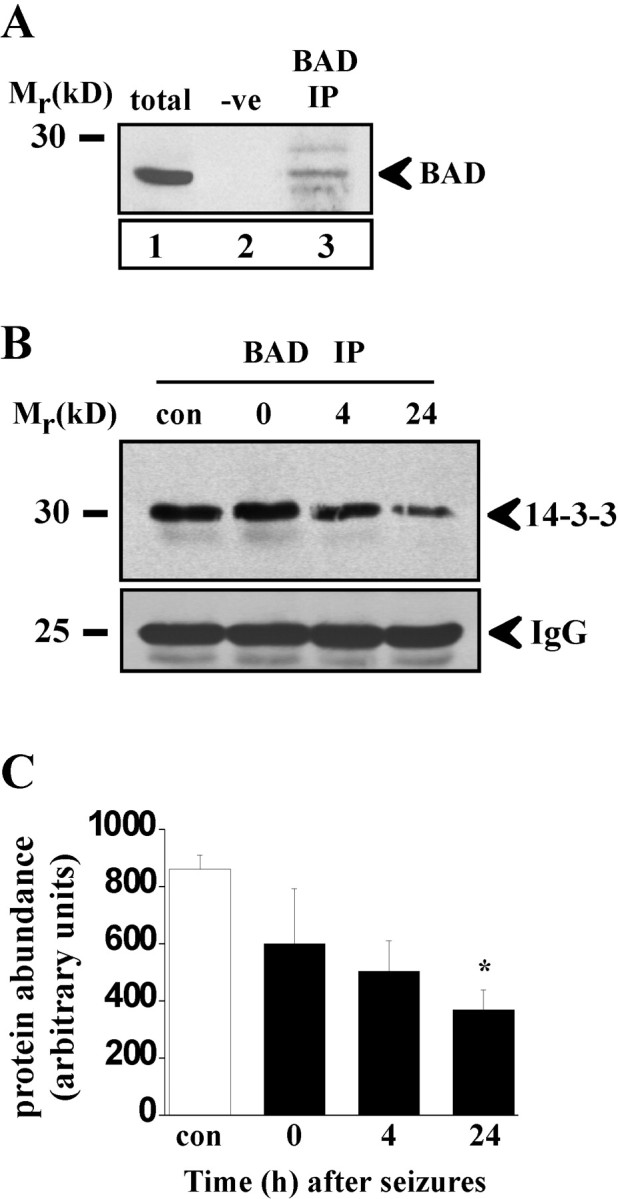
Bcl-2 family gene products are critical to the integration of cell death stimuli that target the mitochondrion. Proapoptotic BAD (Bcl-2-associated death protein) has been shown to dissociate from its sequestered site with the molecular chaperone protein 14-3-3 and displace proapoptotic BAX (Bcl-2-associated X protein) from antiapoptotic BCL-Xl. BAX subsequently translocates to the mitochondrion and induces cytochrome c release and caspase activation. Herein we report the response of the key members of this proposed pathway after seizures. Seizures evoked by microinjection of kainic acid into the amygdala of the rat induced unilateral CA3 pyramidal neuron death with features of apoptosis. In control hippocampus and cortex, BAD was found constitutively bound to 14-3-3, whereas BCL-Xl bound BAX. Within damaged hippocampus, seizures induced the dissociation of BAD from 14-3-3 and the subsequent dimerization of BAD with BCL-Xl as determined by immunoprecipitation and immunohistochemical colocalization. 14-3-3 was found to translocate to the nucleus of degenerating neurons, whereas BAX accumulated at mitochondrial membranes. In contrast, the primarily uninjured cortex exhibited increased phosphorylation of Akt (protein kinase B), which may phosphorylate and inhibit BAD, and no altered binding of BAD to BCL-Xl. Finally, administration of an inhibitor of phosphatidylinositol 3-kinase (LY294002), thought to be an upstream activator of Akt, exacerbated cortical apoptosis after seizures. These data suggest that seizures elicit divergent cell death and survival responses within neuronal populations and that the BAD cell death pathway may perform an instigator or reinforcement role in seizure-induced neuronal death.
Figures







Similar articles
-
Seizure-like activity leads to the release of BAD from 14-3-3 protein and cell death in hippocampal neurons in vitro.Cell Death Differ. 2003 May;10(5):539-47. doi: 10.1038/sj.cdd.4401206. Cell Death Differ. 2003. PMID: 12728252
-
Subcellular distribution of Bcl-2 family proteins and 14-3-3 within the hippocampus during seizure-induced neuronal death in the rat.Neurosci Lett. 2004 Feb 19;356(3):163-6. doi: 10.1016/j.neulet.2003.11.048. Neurosci Lett. 2004. PMID: 15036620
-
Interaction of 14-3-3 with Bid during seizure-induced neuronal death.J Neurochem. 2003 Jul;86(2):460-9. doi: 10.1046/j.1471-4159.2003.01860.x. J Neurochem. 2003. PMID: 12871587
-
Targeting WNT, protein kinase B, and mitochondrial membrane integrity to foster cellular survival in the nervous system.Histol Histopathol. 2004 Apr;19(2):495-504. doi: 10.14670/HH-19.495. Histol Histopathol. 2004. PMID: 15024710 Free PMC article. Review.
-
Oxidative stress and mitochondrial dysfunction as determinants of ischemic neuronal death and survival.J Neurochem. 2009 May;109 Suppl 1(Suppl 1):133-8. doi: 10.1111/j.1471-4159.2009.05897.x. J Neurochem. 2009. PMID: 19393019 Free PMC article. Review.
Cited by
-
Succinate accumulation induces mitochondrial reactive oxygen species generation and promotes status epilepticus in the kainic acid rat model.Redox Biol. 2020 Jan;28:101365. doi: 10.1016/j.redox.2019.101365. Epub 2019 Oct 31. Redox Biol. 2020. PMID: 31707354 Free PMC article.
-
Microglia are mediators of Borrelia burgdorferi-induced apoptosis in SH-SY5Y neuronal cells.PLoS Pathog. 2009 Nov;5(11):e1000659. doi: 10.1371/journal.ppat.1000659. Epub 2009 Nov 13. PLoS Pathog. 2009. PMID: 19911057 Free PMC article.
-
Effect of Nobiletin on Experimental Model of Epilepsy.Transl Neurosci. 2018 Dec 31;9:211-219. doi: 10.1515/tnsci-2018-0031. eCollection 2018. Transl Neurosci. 2018. PMID: 30746285 Free PMC article.
-
Cell signaling underlying epileptic behavior.Front Behav Neurosci. 2011 Aug 2;5:45. doi: 10.3389/fnbeh.2011.00045. eCollection 2011. Front Behav Neurosci. 2011. PMID: 21852968 Free PMC article.
-
Oxidative damage to RNA: mechanisms, consequences, and diseases.Cell Mol Life Sci. 2010 Jun;67(11):1817-29. doi: 10.1007/s00018-010-0277-y. Epub 2010 Feb 11. Cell Mol Life Sci. 2010. PMID: 20148281 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous