

Chk2-deficient mice exhibit radioresistance and defective p53-mediated transcription
- PMID: 12356735
- PMCID: PMC129029
- DOI: 10.1093/emboj/cdf506
Chk2-deficient mice exhibit radioresistance and defective p53-mediated transcription
Abstract
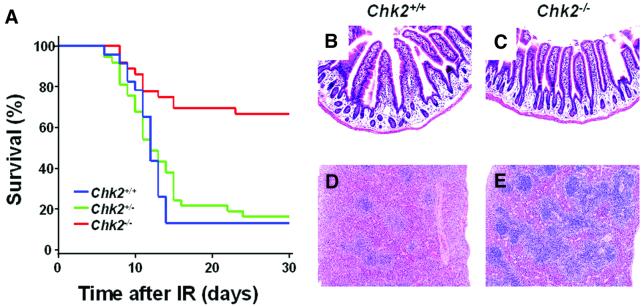
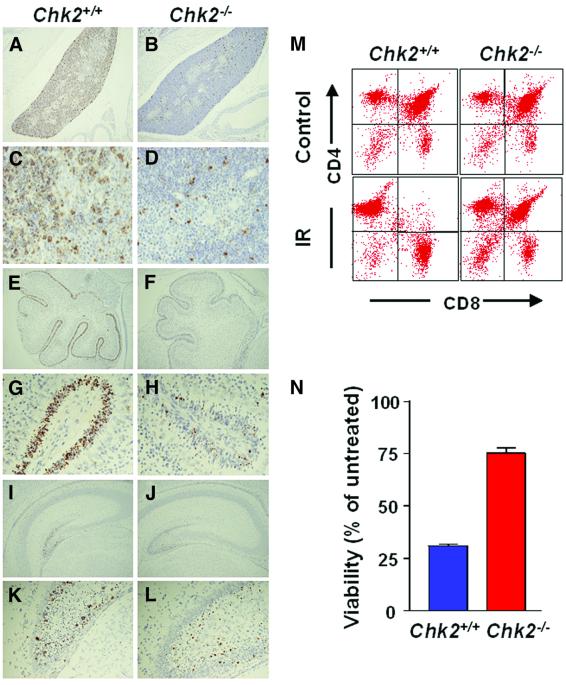
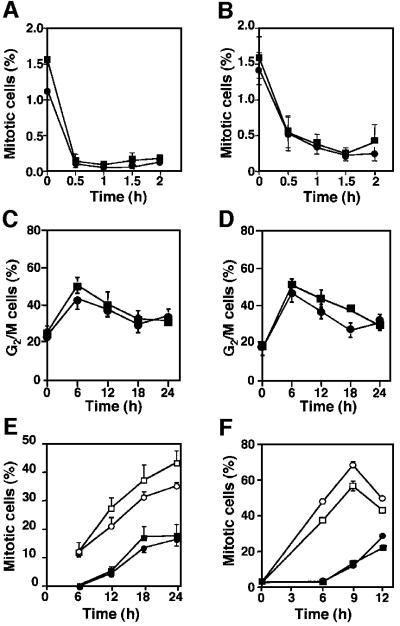
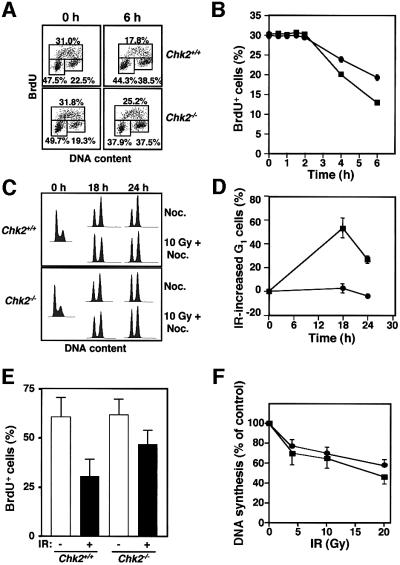
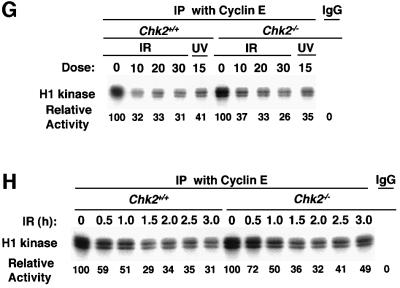
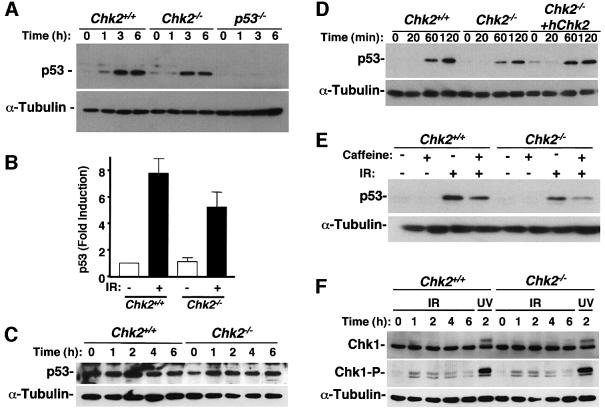
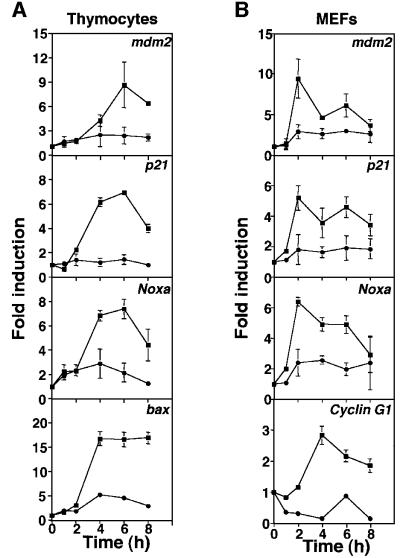
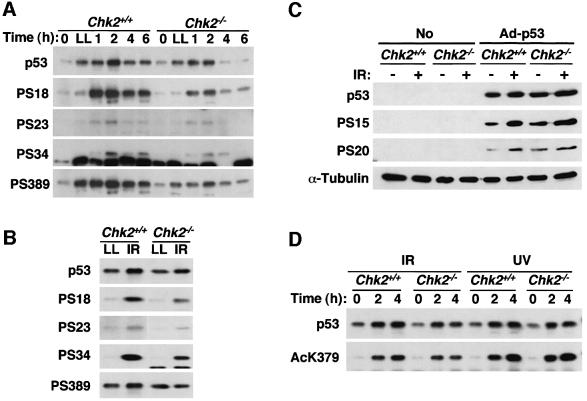
The mammalian Chk2 kinase is thought to mediate ATM-dependent signaling in response to DNA damage. The physiological role of mammalian Chk2 has now been investigated by the generation of Chk2-deficient mice. Although Chk2(-/-) mice appeared normal, they were resistant to ionizing radiation (IR) as a result of the preservation of splenic lymphocytes. Thymocytes and neurons of the developing brain were also resistant to IR-induced apoptosis. The IR-induced G(1)/S cell cycle checkpoint, but not the G(2)/M or S phase checkpoints, was impaired in embryonic fibroblasts derived from Chk2(-/-) mice. IR-induced stabilization of p53 in Chk2(-/- )cells was 50-70% of that in wild-type cells. Caffeine further reduced p53 accumulation, suggesting the existence of an ATM/ATR-dependent but Chk2-independent pathway for p53 stabilization. In spite of p53 protein stabilization and phosphorylation of Ser23, p53-dependent transcriptional induction of target genes, such as p21 and Noxa, was not observed in Chk2(-/-) cells. Our results show that Chk2 plays a critical role in p53 function in response to IR by regulating its transcriptional activity as well as its stability.
Figures
Comment in
-
CHK2: a tumor suppressor or not?Cell Cycle. 2002 Nov-Dec;1(6):401-3. doi: 10.4161/cc.1.6.264. Cell Cycle. 2002. PMID: 12548013 Review. No abstract available.
References
-
- Appella E. and Anderson,C.W. (2001) Post-translational modifications and activation of p53 by genotoxic stresses. Eur. J. Biochem., 268, 2764–2772. - PubMed
-
- Banin S. et al. (1998) Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science, 281, 1674–1677. - PubMed
-
- Barlow C. et al. (1996) Atm-deficient mice: a paradigm of ataxia telangiectasia. Cell, 86, 159–171. - PubMed
-
- Barlow C., Brown,K.D., Deng,C.X., Tagle,D.A. and Wynshaw-Boris,A. (1997) Atm selectively regulates distinct p53-dependent cell-cycle checkpoint and apoptotic pathways. Nat. Genet., 17, 453–456. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
