

Identification and analysis of over 2000 ribosomal protein pseudogenes in the human genome
- PMID: 12368239
- PMCID: PMC187539
- DOI: 10.1101/gr.331902
Identification and analysis of over 2000 ribosomal protein pseudogenes in the human genome
Abstract
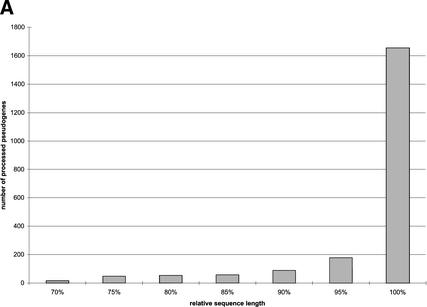
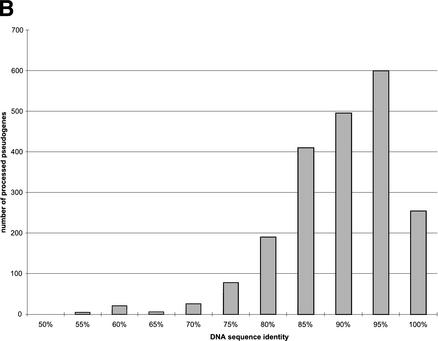
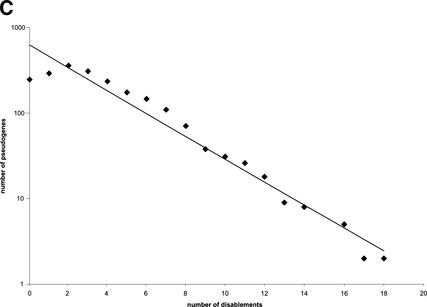
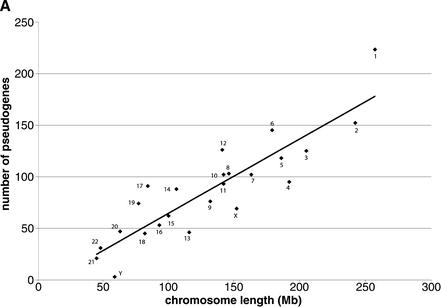
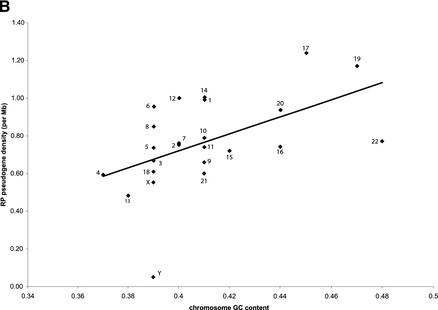
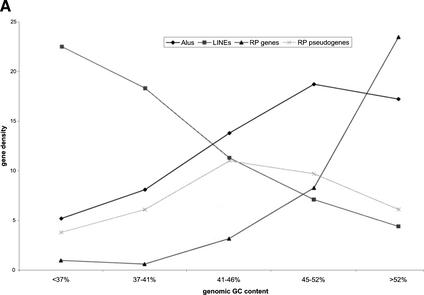
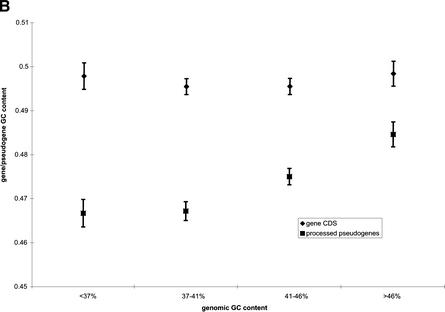
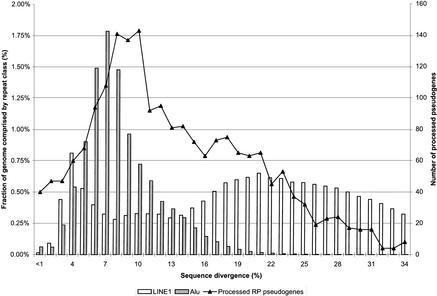
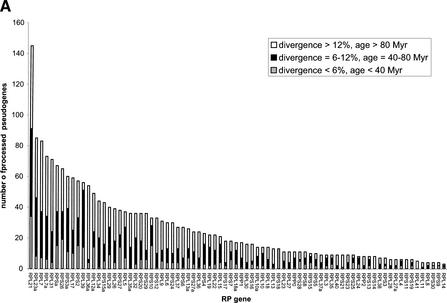
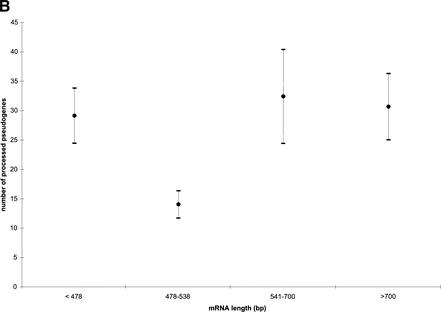
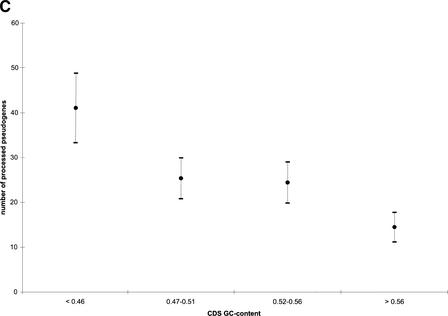
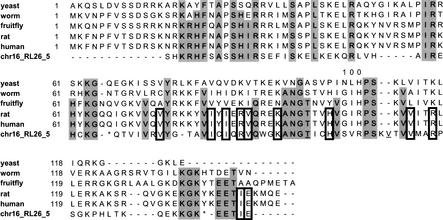
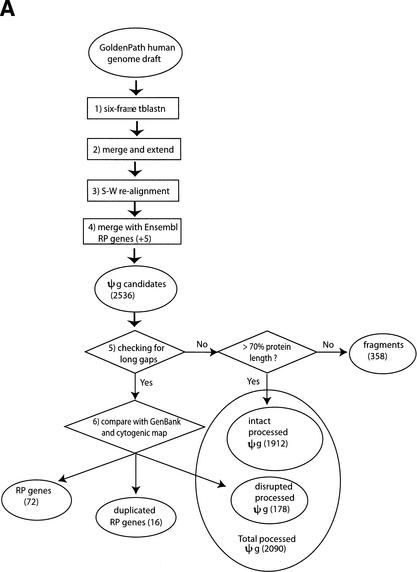
Mammals have 79 ribosomal proteins (RP). Using a systematic procedure based on sequence-homology, we have comprehensively identified pseudogenes of these proteins in the human genome. Our assignments are available at http://www.pseudogene.org or http://bioinfo.mbb.yale.edu/genome/pseudogene. In total, we found 2090 processed pseudogenes and 16 duplications of RP genes. In relation to the matching parent protein, each of the processed pseudogenes has an average relative sequence length of 97% and an average sequence identity of 76%. A small number (258) of them do not contain obvious disablements (stop codons or frameshifts) and, therefore, could be mistaken as functional genes, and 178 are disrupted by one or more repetitive elements. On average, processed pseudogenes have a longer truncation at the 5' end than the 3' end, consistent with the target-primed-reverse-transcription (TPRT) mechanism. Interestingly, on chromosome 16, an RPL26 processed pseudogene was found in the intron region of a functional RPS2 gene. The large-scale distribution of RP pseudogenes throughout the genome appears to result, chiefly, from random insertions with the numbers on each chromosome, consequently, proportional to its size. In contrast to RP genes, the RP pseudogenes have the highest density in GC-intermediate regions (41%-46%) of the genome, with the density pattern being between that of LINEs and Alus. This can be explained by a negative selection theory as we observed that GC-rich RP pseudogenes decay faster in GC-poor regions. Also, we observed a correlation between the number of processed pseudogenes and the GC content of the associated functional gene, i.e., relatively GC-poor RPs have more processed pseudogenes. This ranges from 145 pseudogenes for RPL21 down to 3 pseudogenes for RPL14. We were able to date the RP pseudogenes based on their sequence divergence from present-day RP genes, finding an age distribution similar to that for Alus. The distribution is consistent with a decline in retrotransposition activity in the hominid lineage during the last 40 Myr. We discuss the implications for retrotransposon stability and genome dynamics based on these new findings.
Figures

References
-
- Ban N, Nissen P, Hansen J, Moore PB, Steitz T. The complete atomic structure of the large ribosomal subunit at 2.4 A resolution. Nature. 2000;400:841–847. - PubMed
-
- Bernardi G. Isochores and the evolutionary genomics of vertebrates. Gene. 2000;241:3–17. - PubMed
-
- ————— Misunderstandings about isochores. Part 1. Gene. 2001;276:3–13. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous