

Judging the quality of gene expression-based clustering methods using gene annotation
- PMID: 12368250
- PMCID: PMC187526
- DOI: 10.1101/gr.397002
Judging the quality of gene expression-based clustering methods using gene annotation
Abstract
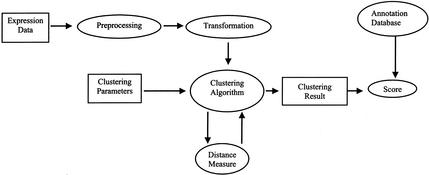
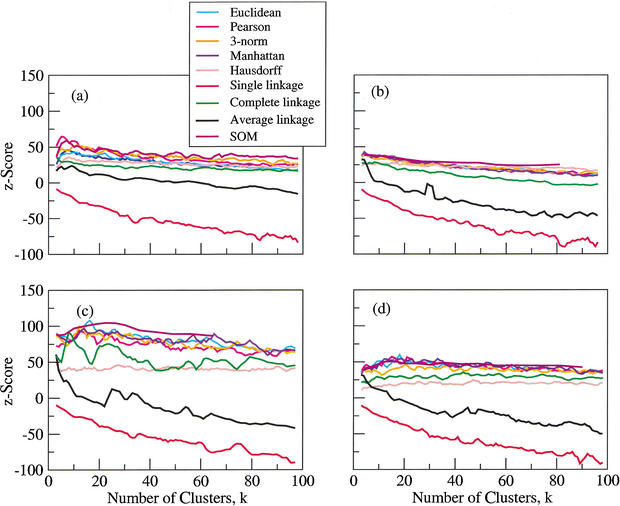
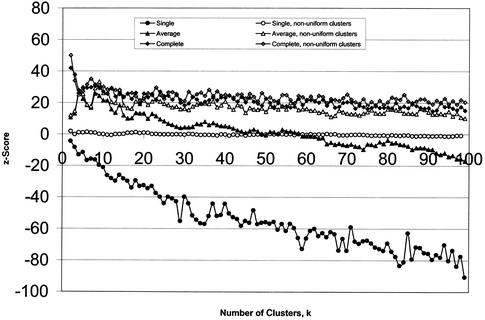
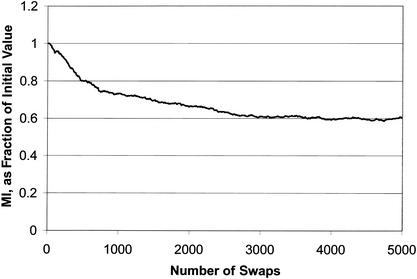
We compare several commonly used expression-based gene clustering algorithms using a figure of merit based on the mutual information between cluster membership and known gene attributes. By studying various publicly available expression data sets we conclude that enrichment of clusters for biological function is, in general, highest at rather low cluster numbers. As a measure of dissimilarity between the expression patterns of two genes, no method outperforms Euclidean distance for ratio-based measurements, or Pearson distance for non-ratio-based measurements at the optimal choice of cluster number. We show the self-organized-map approach to be best for both measurement types at higher numbers of clusters. Clusters of genes derived from single- and average-linkage hierarchical clustering tend to produce worse-than-random results.
Figures
References
-
- Aach J, Rindone W, Church GM. Systematic management and analysis of yeast gene expression data. Genome Res. 2000;10:431–445. - PubMed
-
- Angelo M. GeneCluster. Cambridge, MA: Whitehead/MIT Center for Genome Research; 1999. http://www.genome.wi.mit.edu/cancer/software/software.html ; http://www.genome.wi.mit.edu/cancer/software/software.html. .
-
- Beazley DM. SWIG User's Manual v.1.3; 2001. http://www.swig.org http://www.swig.org. .
-
- Beazley DM, Fletcher D, Dumont D. O'Reilly Perl Conference 2.0. 1998. Perl extension building with SWIG.http://www.swig.org/papers/Per198/swigperl.pdf San Jose, CA; http://www.swig.org/papers/Per198/swigperl.pdf. .
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials