

Role of RANKL and RANK in bone loss and arthritis
- PMID: 12379618
- PMCID: PMC1766717
- DOI: 10.1136/ard.61.suppl_2.ii32
Role of RANKL and RANK in bone loss and arthritis
Abstract
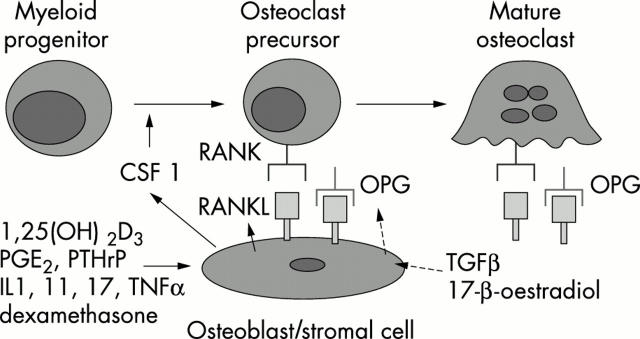
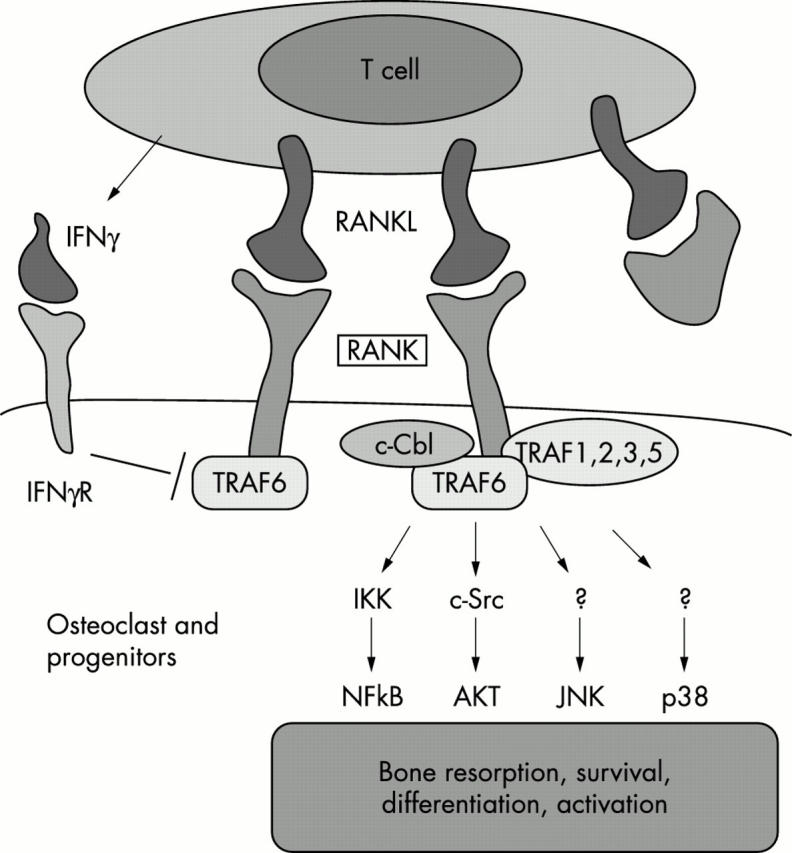
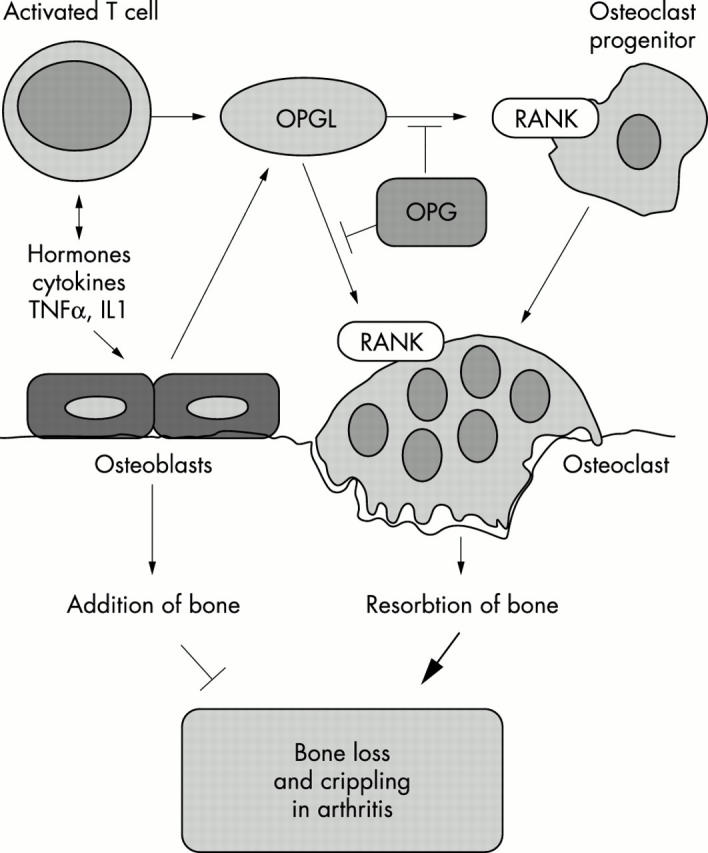
The tumour necrosis factor family molecule RANKL (RANKL, TRANCE, ODF) and its receptor RANK are key regulators of bone remodelling and regulate T cell/dendritic cell communications, and lymph node formation. Moreover, RANKL and RANK are expressed in mammary gland epithelial cells and control the development of a lactating mammary gland during pregnancy and the propagation of mammalian species. Importantly, RANKL and RANK are essential for the development and activation of osteoclasts and bone loss in response to virtually all triggers tested. Therapeutically, inhibition of RANKL function via the decoy receptor osteoprotegerin completely prevents bone loss at inflammed joints and has partially beneficial effects on cartilage destruction in all arthritis models studied. Modulation of these systems provides a unique opportunity to design novel treatments to inhibit bone loss and crippling in arthritis.
Figures
Similar articles
-
RANKL and RANK as novel therapeutic targets for arthritis.Curr Opin Rheumatol. 2003 May;15(3):280-7. doi: 10.1097/00002281-200305000-00016. Curr Opin Rheumatol. 2003. PMID: 12707582 Review.
-
RANK-L and RANK: T cells, bone loss, and mammalian evolution.Annu Rev Immunol. 2002;20:795-823. doi: 10.1146/annurev.immunol.20.100301.064753. Epub 2001 Oct 4. Annu Rev Immunol. 2002. PMID: 11861618 Review.
-
Bone destruction in arthritis.Ann Rheum Dis. 2002 Nov;61 Suppl 2(Suppl 2):ii84-6. doi: 10.1136/ard.61.suppl_2.ii84. Ann Rheum Dis. 2002. PMID: 12379632 Free PMC article. Review.
-
Osteoprotegerin and receptor activator of nuclear factor kappaB ligand (RANKL) regulate osteoclast formation by cells in the human rheumatoid arthritic joint.Rheumatology (Oxford). 2001 Jun;40(6):623-30. doi: 10.1093/rheumatology/40.6.623. Rheumatology (Oxford). 2001. PMID: 11426018
-
Clinical implications of the osteoprotegerin/RANKL/RANK system for bone and vascular diseases.JAMA. 2004 Jul 28;292(4):490-5. doi: 10.1001/jama.292.4.490. JAMA. 2004. PMID: 15280347 Review.
Cited by
-
Development of Metabolic Syndrome Decreases Bone Mineral Density T-Score of Calcaneus in Foot in a Large Taiwanese Population Follow-Up Study.J Pers Med. 2021 May 20;11(5):439. doi: 10.3390/jpm11050439. J Pers Med. 2021. PMID: 34065445 Free PMC article.
-
PBT-6, a Novel PI3KC2γ Inhibitor in Rheumatoid Arthritis.Biomol Ther (Seoul). 2020 Mar 1;28(2):172-183. doi: 10.4062/biomolther.2019.153. Biomol Ther (Seoul). 2020. PMID: 31739383 Free PMC article.
-
Exosomal Release of L-Plastin by Breast Cancer Cells Facilitates Metastatic Bone Osteolysis.Transl Oncol. 2019 Mar;12(3):462-474. doi: 10.1016/j.tranon.2018.11.014. Epub 2018 Dec 21. Transl Oncol. 2019. PMID: 30583289 Free PMC article.
-
The association between RANK, RANKL and OPG gene polymorphisms and the risk of rheumatoid arthritis: a case-controlled study and meta-analysis.Biosci Rep. 2019 Jun 28;39(6):BSR20182356. doi: 10.1042/BSR20182356. Print 2019 Jun 28. Biosci Rep. 2019. PMID: 31209146 Free PMC article.
-
Association of OPG gene polymorphisms with the risk of knee osteoarthritis among Chinese people.Mol Genet Genomic Med. 2019 Jun;7(6):e662. doi: 10.1002/mgg3.662. Epub 2019 Apr 11. Mol Genet Genomic Med. 2019. PMID: 30972968 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
