

The origin and neuronal function of in vivo nonsynaptic glutamate
- PMID: 12388621
- PMCID: PMC6757683
- DOI: 10.1523/JNEUROSCI.22-20-09134.2002
The origin and neuronal function of in vivo nonsynaptic glutamate
Abstract
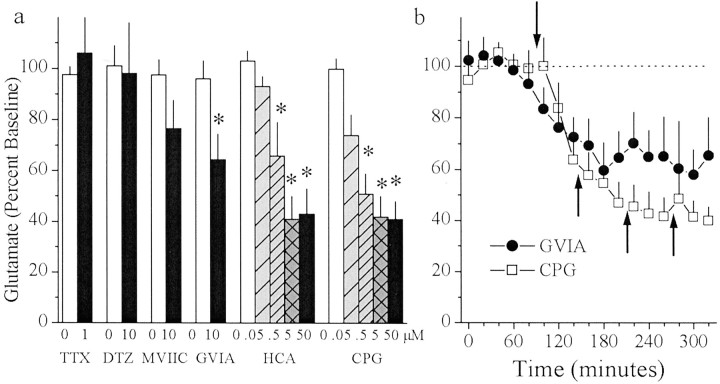
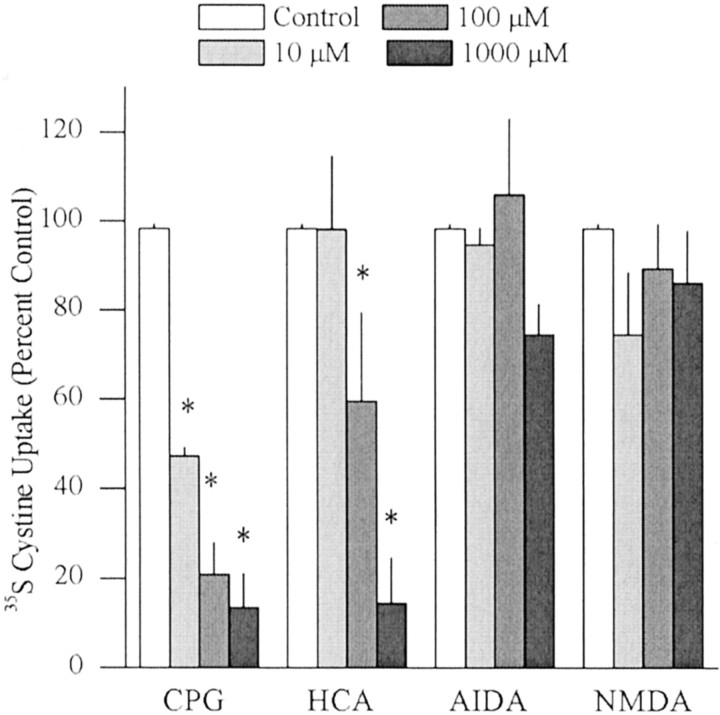
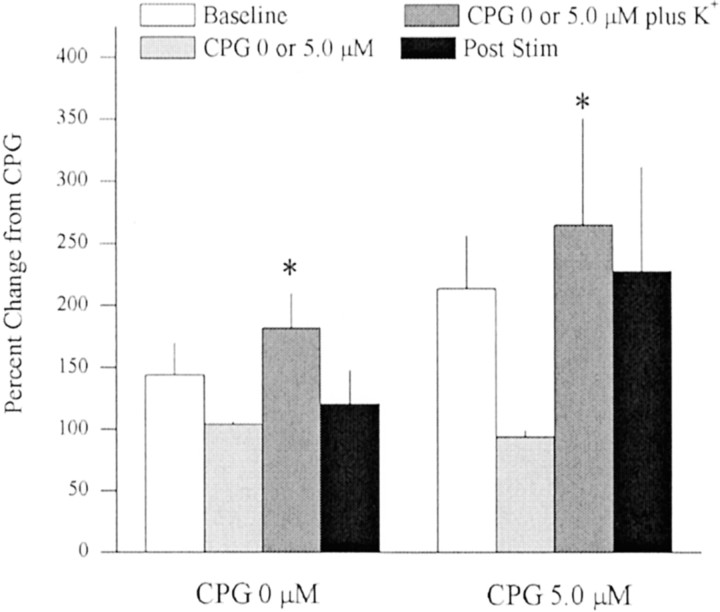
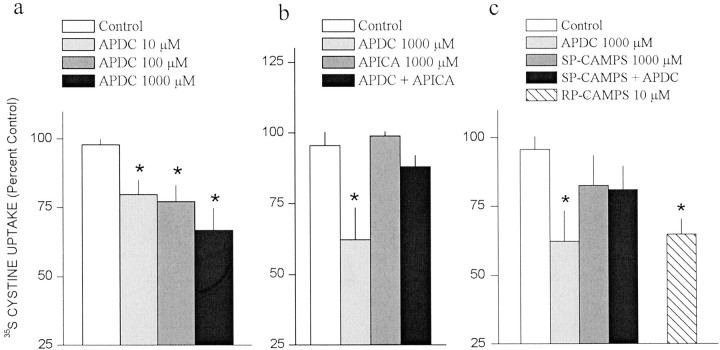
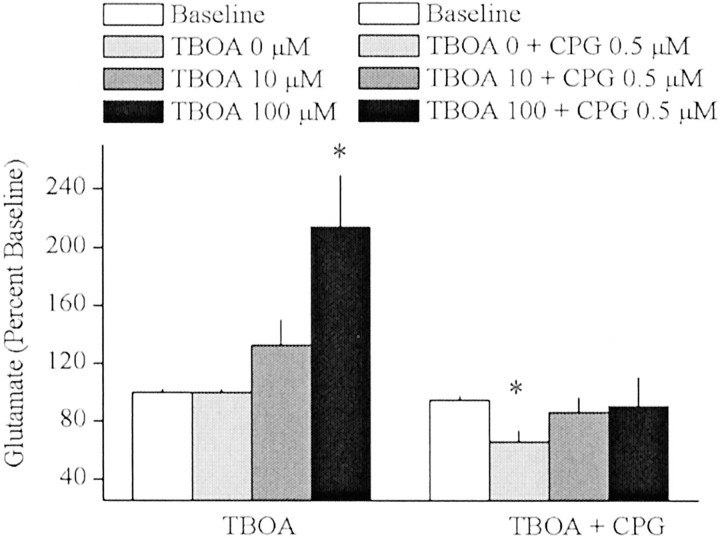
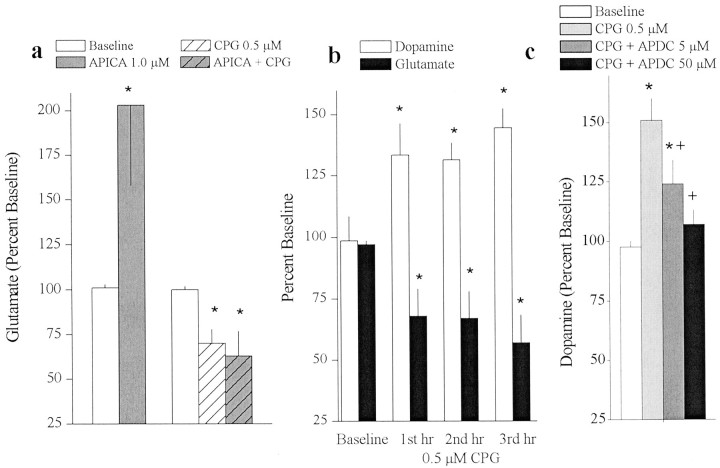
Basal extracellular glutamate sampled in vivo is present in micromolar concentrations in the extracellular space outside the synaptic cleft, and neither the origin nor the function of this glutamate is known. This report reveals that blockade of glutamate release from the cystine-glutamate antiporter produced a significant decrease (60%) in extrasynaptic glutamate levels in the rat striatum, whereas blockade of voltage-dependent Na+ and Ca2+ channels produced relatively minimal changes (0-30%). This indicates that the primary origin of in vivo extrasynaptic glutamate in the striatum arises from nonvesicular glutamate release by the cystine-glutamate antiporter. By measuring [35S]cystine uptake, it was shown that similar to vesicular release, the activity of the cystine-glutamate antiporter is negatively regulated by group II metabotropic glutamate receptors (mGluR2/3) via a cAMP-dependent protein kinase mechanism. Extracellular glutamate derived from the antiporter was shown to regulate extracellular levels of glutamate and dopamine. Infusion of the mGluR2/3 antagonist (RS)-1-amino-5-phosphonoindan-1-carboxylic acid (APICA) increased extracellular glutamate levels, and previous blockade of the antiporter prevented the APICA-induced rise in extracellular glutamate. This suggests that glutamate released from the antiporter is a source of endogenous tone on mGluR2/3. Blockade of the antiporter also produced an increase in extracellular dopamine that was reversed by infusing the mGluR2/3 agonist (2R,4R)-4-aminopyrrolidine-2,4-dicarboxlylate, indicating that antiporter-derived glutamate can modulate dopamine transmission via mGluR2/3 heteroreceptors. These results suggest that nonvesicular release from the cystine-glutamate antiporter is the primary source of in vivo extracellular glutamate and that this glutamate can modulate both glutamate and dopamine transmission.
Figures
References
-
- Alagarsamy S, Sorensen SD, Conn PJ. Coordinate regulation of metabotropic glutamate receptors. Curr Opin Neurobiol. 2001;11:357–362. - PubMed
-
- Anwyl R. Metabotropic glutamate receptors: electrophysiological properties and role in plasticity. Brain Res Rev. 1999;29:83–120. - PubMed
-
- Bannai S. Exchange of cystine and glutamate across plasma membrane of human fibroblasts. J Biol Chem. 1986;261:2256–2263. - PubMed
-
- Bannai S, Ishii T. Transport of cystine and cysteine and cell growth in cultured human diploid fibroblasts: effect of glutamate and homocysteate. J Cell Physiol. 1982;112:265–272. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous