

Activation of the mouse histone deacetylase 1 gene by cooperative histone phosphorylation and acetylation
- PMID: 12391151
- PMCID: PMC134744
- DOI: 10.1128/MCB.22.22.7820-7830.2002
Activation of the mouse histone deacetylase 1 gene by cooperative histone phosphorylation and acetylation
Abstract
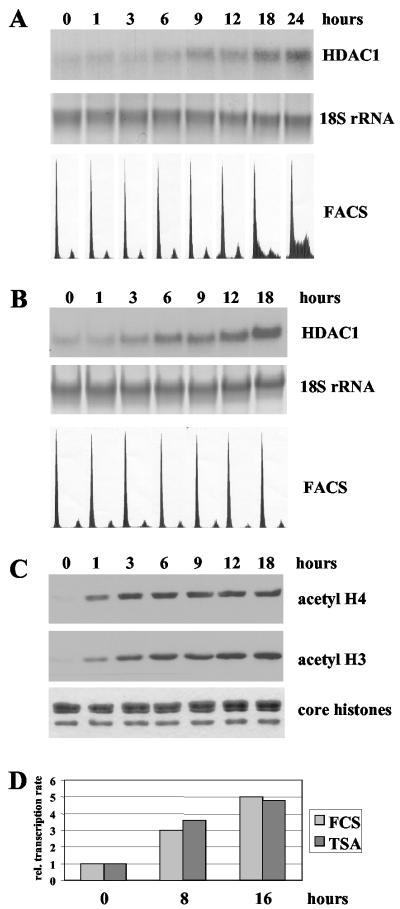
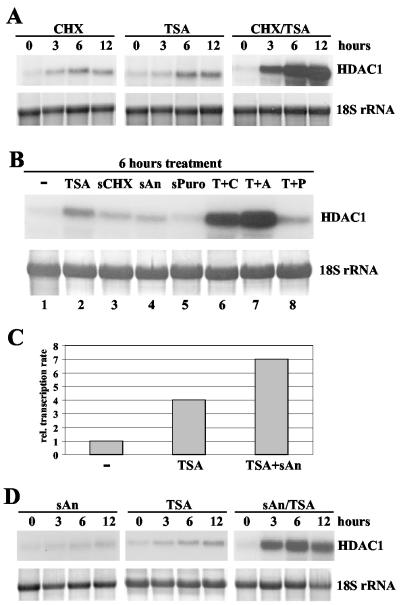
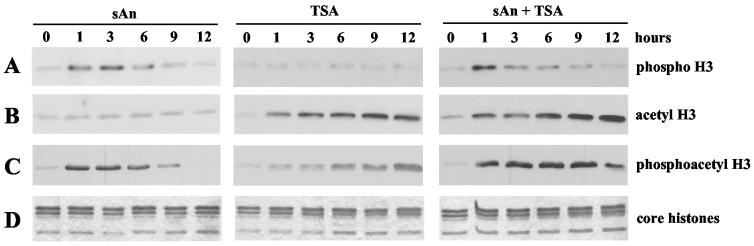
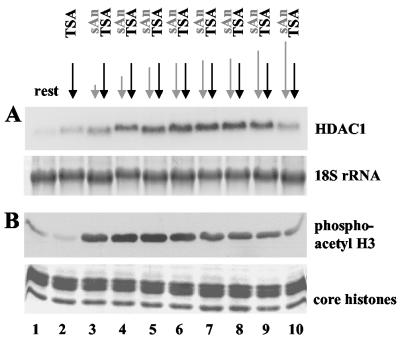
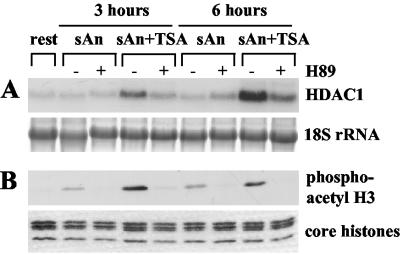
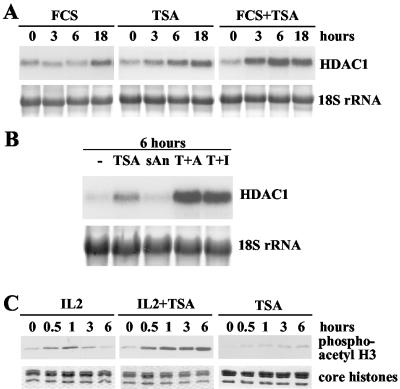
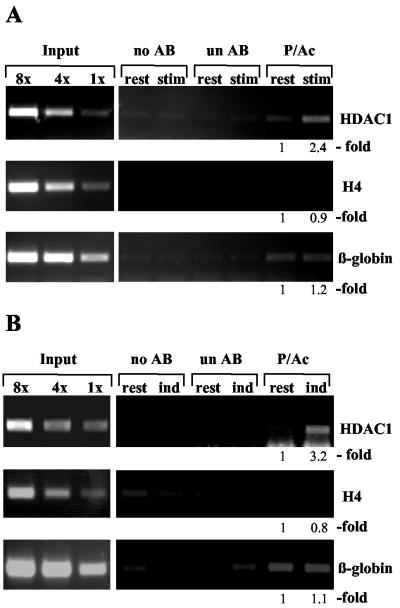
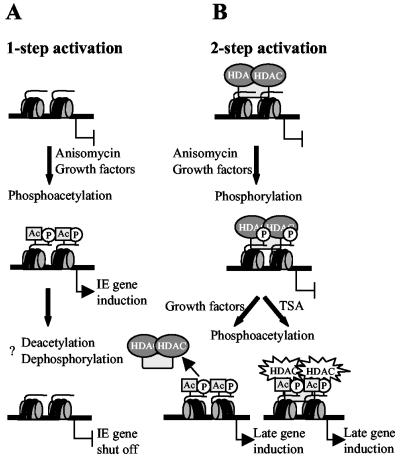
Histone deacetylase 1 (HDAC1) is a major regulator of chromatin structure and gene expression. Tight control of HDAC1 expression is essential for normal cell cycle progression of mammalian cells. HDAC1 mRNA levels are regulated by growth factors and by changes in intracellular deacetylase activity levels. Stimulation of the mitogen-activated protein kinase cascade by anisomycin or growth factors, together with inhibition of deacetylases by trichostatin A (TSA), leads to stable histone H3 phosphoacetylation and strongly induced HDAC1 expression. In contrast, activation of the nucleosomal response by anisomycin alone results only in transient phosphoacetylation of histone H3 without affecting HDAC1 mRNA levels. The transcriptional induction of the HDAC1 gene by anisomycin and TSA is efficiently blocked by H89, an inhibitor of the nucleosomal response. Detailed studies of the kinetics of histone acetylation and phosphorylation show that the two modifications are synergistic and essential for induced HDAC1 transcription. Activation of the HDAC1 gene by anisomycin together with TSA or by growth factors is accompanied by phosphoacetylation of HDAC1 promoter-associated histone H3. Our results present evidence for a precise regulatory mechanism which allows induction of the HDAC1 gene in response to proliferation signals and modulation of HDAC1 expression dependent on intracellular deacetylase levels.
Figures
Similar articles
-
Autoregulation of mouse histone deacetylase 1 expression.Mol Cell Biol. 2003 Oct;23(19):6993-7004. doi: 10.1128/MCB.23.19.6993-7004.2003. Mol Cell Biol. 2003. PMID: 12972616 Free PMC article.
-
Trichostatin A induces 5-lipoxygenase promoter activity and mRNA expression via inhibition of histone deacetylase 2 and 3.J Cell Mol Med. 2012 Jul;16(7):1461-73. doi: 10.1111/j.1582-4934.2011.01420.x. J Cell Mol Med. 2012. PMID: 21883892 Free PMC article.
-
Inhibitors of histone deacetylase and DNA methyltransferase synergistically activate the methylated metallothionein I promoter by activating the transcription factor MTF-1 and forming an open chromatin structure.Mol Cell Biol. 2002 Dec;22(23):8302-19. doi: 10.1128/MCB.22.23.8302-8319.2002. Mol Cell Biol. 2002. PMID: 12417732 Free PMC article.
-
Assessment of Synergistic Contribution of Histone Deacetylases in Prognosis and Therapeutic Management of Sarcoma.Mol Diagn Ther. 2020 Oct;24(5):557-569. doi: 10.1007/s40291-020-00487-2. Mol Diagn Ther. 2020. PMID: 32696211 Review.
-
HDAC1: an environmental sensor regulating endothelial function.Cardiovasc Res. 2022 Jun 29;118(8):1885-1903. doi: 10.1093/cvr/cvab198. Cardiovasc Res. 2022. PMID: 34264338 Free PMC article. Review.
Cited by
-
Desmin enters the nucleus of cardiac stem cells and modulates Nkx2.5 expression by participating in transcription factor complexes that interact with the nkx2.5 gene.Biol Open. 2016 Jan 19;5(2):140-53. doi: 10.1242/bio.014993. Biol Open. 2016. PMID: 26787680 Free PMC article.
-
Down-regulation of matrix metalloproteinase-7 inhibits metastasis of human anaplastic thyroid cancer cell line.Clin Exp Metastasis. 2012 Jan;29(1):71-82. doi: 10.1007/s10585-011-9430-8. Epub 2011 Nov 1. Clin Exp Metastasis. 2012. PMID: 22042554
-
Tissue specific regulation of the chick Sox10E1 enhancer by different Sox family members.Dev Biol. 2017 Feb 1;422(1):47-57. doi: 10.1016/j.ydbio.2016.12.004. Epub 2016 Dec 22. Dev Biol. 2017. PMID: 28012818 Free PMC article.
-
Divergent roles of HDAC1 and HDAC2 in the regulation of epidermal development and tumorigenesis.EMBO J. 2013 Dec 11;32(24):3176-91. doi: 10.1038/emboj.2013.243. Epub 2013 Nov 15. EMBO J. 2013. PMID: 24240174 Free PMC article.
-
A downstream CpG island controls transcript initiation and elongation and the methylation state of the imprinted Airn macro ncRNA promoter.PLoS Genet. 2012;8(3):e1002540. doi: 10.1371/journal.pgen.1002540. Epub 2012 Mar 1. PLoS Genet. 2012. PMID: 22396659 Free PMC article.
References
-
- Chadee, D. N., M. J. Hendzel, C. P. Tylipski, C. D. Allis, D. P. Bazett Jones, J. A. Wright, and J. R. Davie. 1999. Increased Ser-10 phosphorylation of histone H3 in mitogen-stimulated and oncogene-transformed mouse fibroblasts. J. Biol. Chem. 274:24914-24920. - PubMed
-
- Cheung, P., C. D. Allis, and P. Sassone-Corsi. 2000. Signaling to chromatin through histone modifications. Cell 103:263-271. - PubMed
-
- Cheung, P., K. G. Tanner, W. L. Cheung, P. Sassone Corsi, J. M. Denu, and C. D. Allis. 2000. Synergistic coupling of histone H3 phosphorylation and acetylation in response to epidermal growth factor stimulation. Mol. Cell 5:905-915. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous