

Review
doi: 10.1086/344412.
Epub 2002 Oct 22.
Multiple hits during early embryonic development: digenic diseases and holoprosencephaly
Affiliations
- PMID: 12395298
- PMCID: PMC385082
- DOI: 10.1086/344412
Item in Clipboard
Review
Multiple hits during early embryonic development: digenic diseases and holoprosencephaly
Am J Hum Genet.
2002 Nov.
No abstract available
Figures
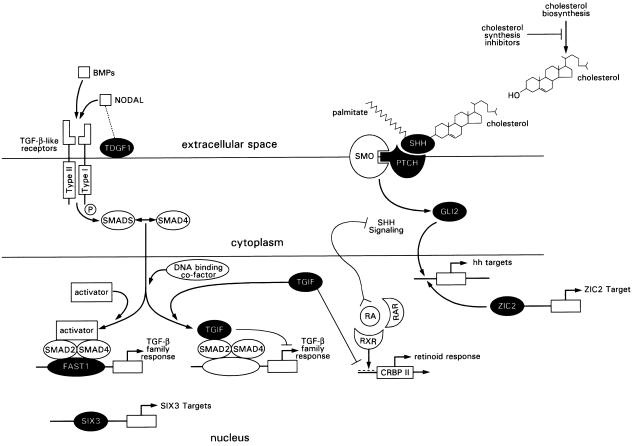
Association of HPE with abnormal signaling pathways and cholesterol biosynthesis and with ZIC2 and SIX3 transcription factors. Some aspects of interactions are speculative, since not all links have been explicitly demonstrated experimentally and many of the links are only suggestive. In addition, results have been synthesized from several species although they are depicted here for humans only. To date, the following genes (blackened symbols) have been implicated in HPE in humans: SHH, PTCH, GLI2 in Sonic hedgehog signaling, TDGF1, FAST1, TGIF in Nodal/TGF-β signaling, SIX3, and ZIC2 (updated and modified from Muenke and Beachy 2001).
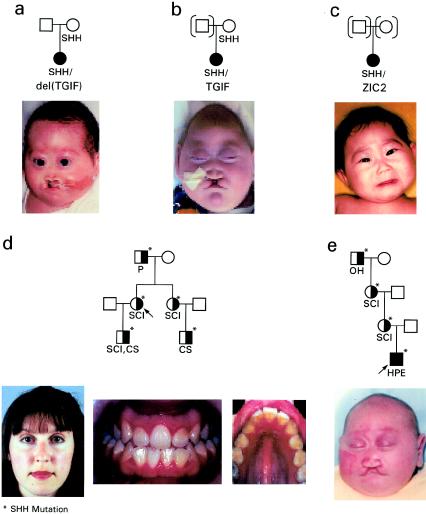
Pedigrees segregating different missense mutations in SHH. A–C, Patients with HPE and mutations in a second gene. D and E, Individuals with HPE microsign (CS = choanal stenosis; P = ptosis of eyelids; OH = ocular hypotelorism) are depicted by half-blackened symbols. One pedigree segregates an SHH (I111F) mutation (modified from Nanni et al. 2001) (D). One pedigree segregates an SHH (S236R) mutation (modified from Nanni et al. 1999) (E). Patient photographs have been published elsewhere (Nanni et al. [patients in panels A, C, and E], Nanni et al. [patient in panel D]; Gripp et al. [patient in panel B]) and are used with permission of the publishers.
References
Electronic-Database Information
-
- Online Mendelian Inheritance in Man (OMIM) http://www.ncbi.nlm.nih.gov/Omim/ (for ADPKD [MIM #173900], Bardet-Biedl Syndrome [MIM #209900], congenital glaucoma [MIM #231300], early onset glaucoma [MIM #137750], GABEB [MIM #226650], JEB [MIM #226700], ocular albinism [MIM #203100], RDS [MIM #179605], USH1B [MIM #276903], USH3 [MIM #276902], and WS2 [MIM #193510])
References
-
- Alward WL, Fingert JH, Coote MA, Johnson AT, Lerner SF, Junqua D, Durcan FJ, McCartney PJ, Mackey DA, Sheffield VC, Stone EM (1998) Clinical features associated with mutations in the chromosome 1 open-angle glaucoma gene (GLC1A). N Engl J Med 338:1022–1027 - PubMed
-
- Angrist M, Bolk S, Halushka M, Lapchak PA, Chakravarti A (1996) Germline mutations in glial cell line-derived neurotrophic factor (GDNF) and RET in a Hirschsprung disease patient. Nat Genet 14:341–344 - PubMed
-
- Aruga J, Yokota N, Hashimoto M, Furuichi T, Fukuda M, Mikoshiba K (1994) A novel zinc finger protein, zic, is involved in neurogenesis, especially in the cell lineage of cerebellar granule cells. J Neurochem 63:1880–1890 - PubMed
-
- Auricchio A, Griseri P, Carpentieri ML, Betsos N, Staiano A, Tozzi A, Priolo M, Thompson H, Bocciardi R, Romeo G, Ballabio A, Ceccherini I (1999) Double heterozygosity for a RET substitution interfering with splicing and an EDNRB missense mutation in Hirschsprung disease. Am J Hum Genet 64:1216–1221 - PMC - PubMed
Publication types
MeSH terms
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
