

Human L1 element target-primed reverse transcription in vitro
- PMID: 12411507
- PMCID: PMC131089
- DOI: 10.1093/emboj/cdf592
Human L1 element target-primed reverse transcription in vitro
Abstract
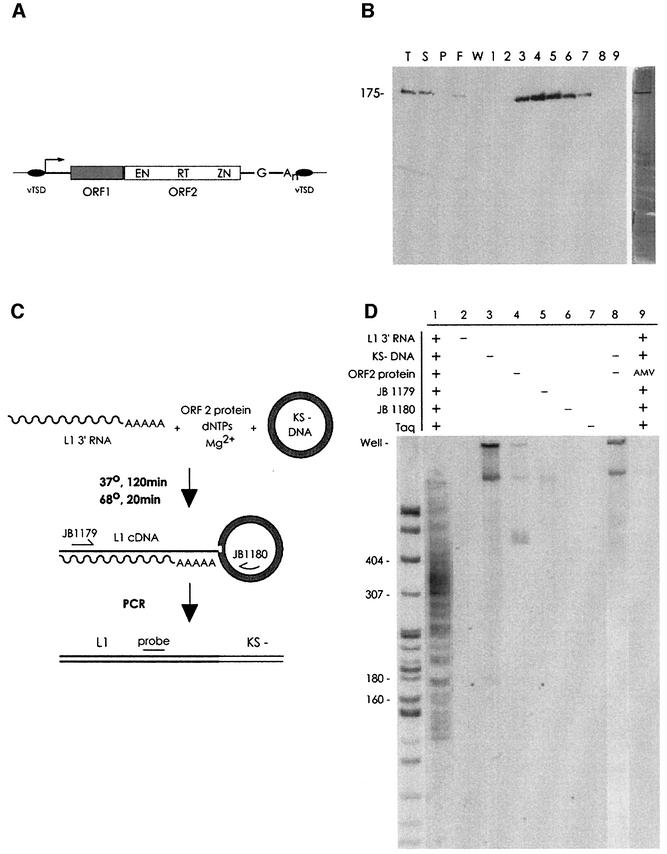
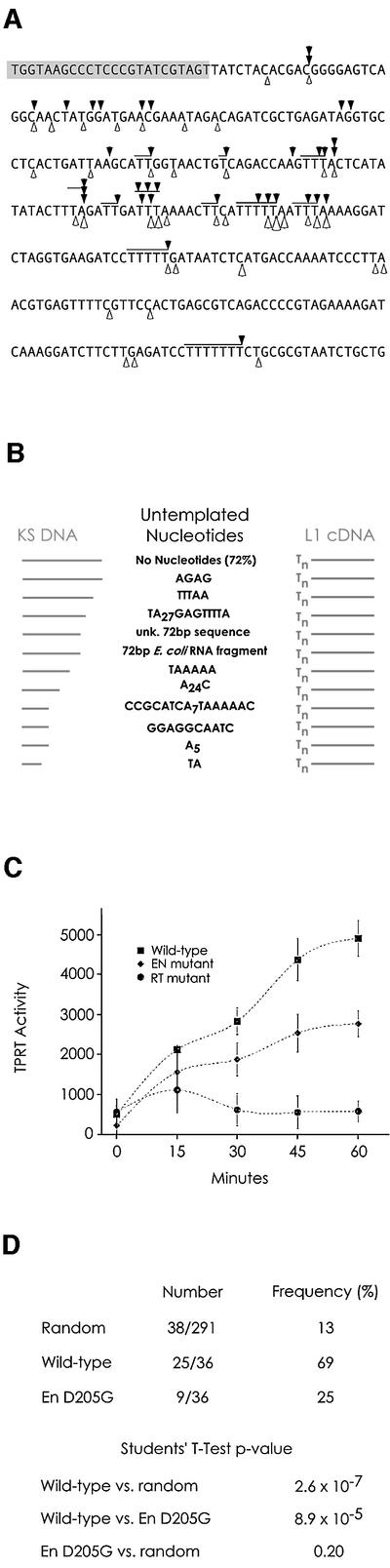
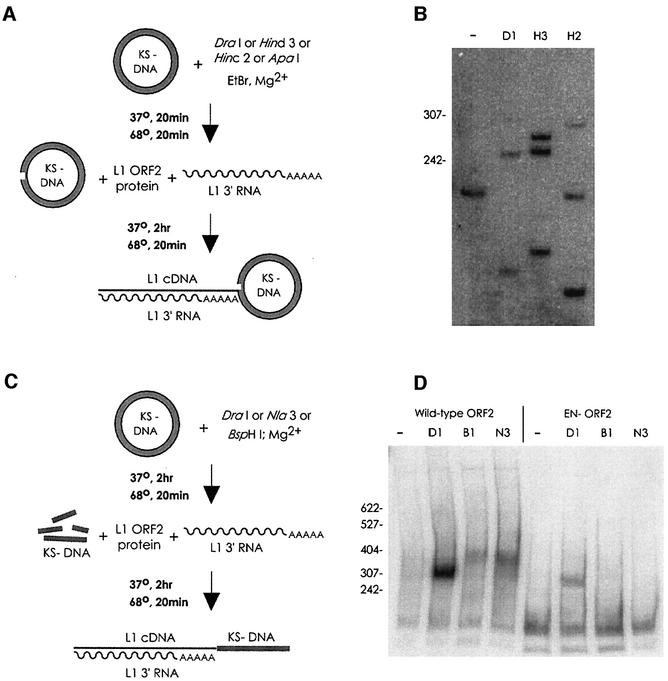
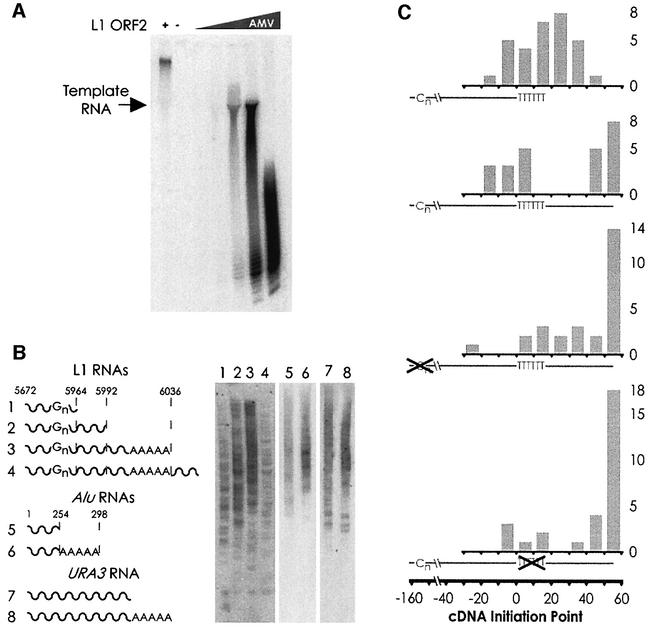
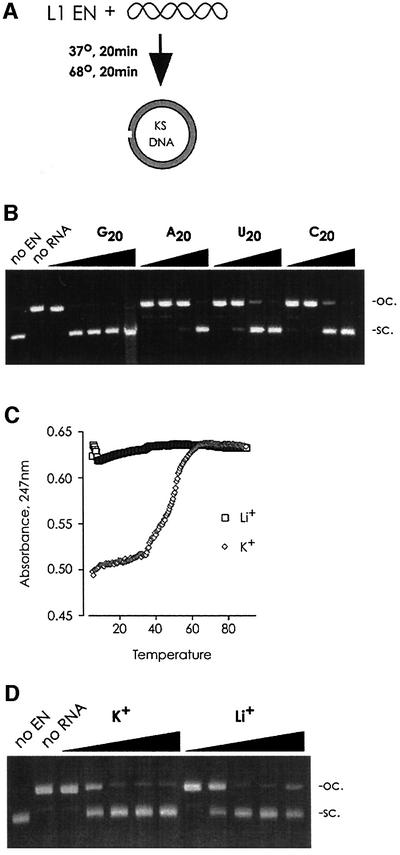
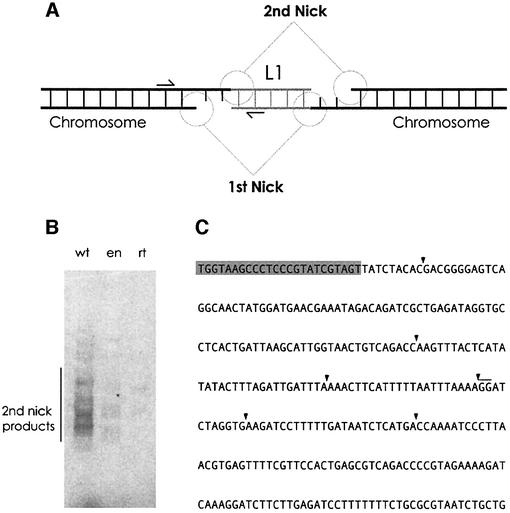
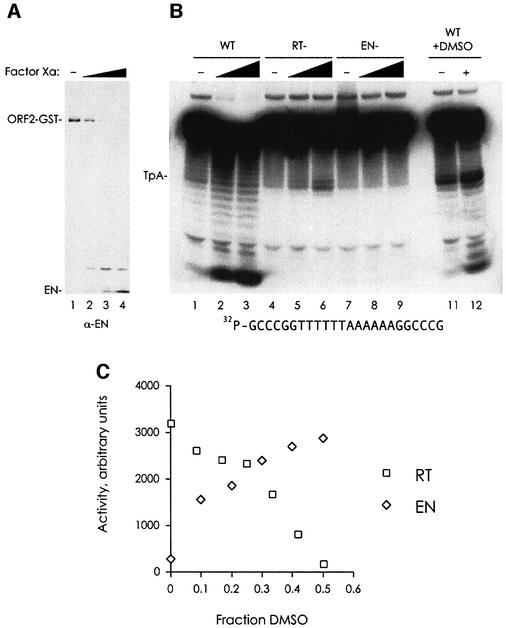
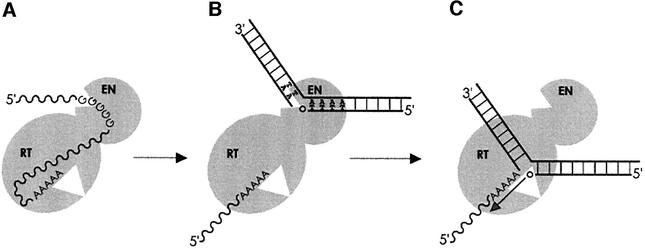
L1 elements are ubiquitous human transposons that replicate via an RNA intermediate. We have reconstituted the initial stages of L1 element transposition in vitro. The reaction requires only the L1 ORF2 protein, L1 3' RNA, a target DNA and appropriate buffer components. We detect branched molecules consisting of junctions between transposon 3' end cDNA and the target DNA, resulting from priming at a nick in the target DNA. 5' junctions of transposon cDNA and target DNA are also observed. The nicking and reverse transcription steps in the reaction can be uncoupled, as priming at pre-existing nicks and even double-strand breaks can occur. We find evidence for specific positioning of the L1 RNA with the ORF2 protein, probably mediated in part by the polyadenosine portion of L1 RNA. Polyguanosine, similar to a conserved region of the L1 3' UTR, potently inhibits L1 endonuclease (L1 EN) activity. L1 EN activity is also repressed in the context of the full-length ORF2 protein, but it and a second cryptic nuclease activity are released by ORF2p proteolysis. Additionally, heterologous RNA species such as Alu element RNA and L1 transcripts with 3' extensions are substrates for the reaction.
Figures
References
-
- Bibillo A. and Eickbush,T.H. (2002) The reverse transcriptase of the R2 non-LTR retrotransposon: continuous synthesis of cDNA on non-continuous RNA templates. J. Mol. Biol., 316, 459–473. - PubMed
-
- Boeke J.D. (1997) LINEs and Alus: the polyA connection. Nat. Genet., 16, 6–7. - PubMed
-
- Boeke J.D. and Stoye,J.P. (1997) Retrotransposons, endogenous retro viruses and the evolution of retroelements. In Coffin,J.M., Hughes,S.H. and Varmus,H.E. (eds), Retroviruses. Cold Spring Harbor Laboratory Press, Plainview, NY, pp. 343–435. - PubMed
-
- Boissinot S., Entezam,A. and Furano,A.V. (2001) Selection against deleterious line-1-containing loci in the human lineage. Mol. Biol. Evol., 18, 926–935. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
