

Review
doi: 10.1172/JCI16781.
Protein aggregation in disease: a role for folding intermediates forming specific multimeric interactions
Affiliations
- PMID: 12417558
- PMCID: PMC151620
- DOI: 10.1172/JCI16781
Item in Clipboard
Review
Protein aggregation in disease: a role for folding intermediates forming specific multimeric interactions
J Clin Invest.
2002 Nov.
No abstract available
Figures
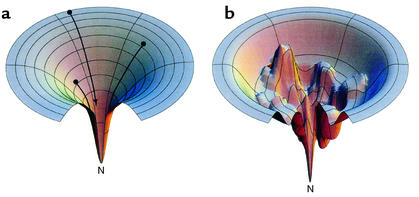
Funnels representing an idealized energy landscape for protein folding (a) or a rugged energy landscape with kinetic traps (b), from models of Dill and Chan (80). (a) The unfolded chain starts at the top of an energy landscape, and, as it forms intrachain contacts, lowering its free energy (and descending the funnel), the number of conformations it can sample is progressively reduced, thus eliminating the need for a global search. Ultimately, it reaches the unique native state at the energetic minimum. Note that the number of pathways typically available coming down from the unfolded state may not be unlimited, as depicted here, but may be multiple. (b) Descending a rugged landscape, the polypeptide can lodge in a kinetic trap or can alternatively descend through a narrow path to the native state. Molecular chaperones may act to “smooth” rugged landscapes, preventing polypeptide from descending into a kinetic trap, or rescuing it from one. Reproduced with permission from Nature Structural Biology (80). N, native state.
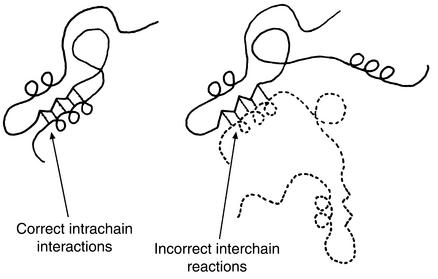
Model of Goldberg and coworkers for protein aggregation involving specific structural contacts formed between two polypeptide chains instead of within a single chain (16). The same bonds are formed but promote incorrect quaternary associations. Kinetic competition between these interchain and intrachain contacts is concentration-dependent, and such contacts are formed from intermediate states (see text). Reproduced with permission from European Journal of Biochemistry (16).
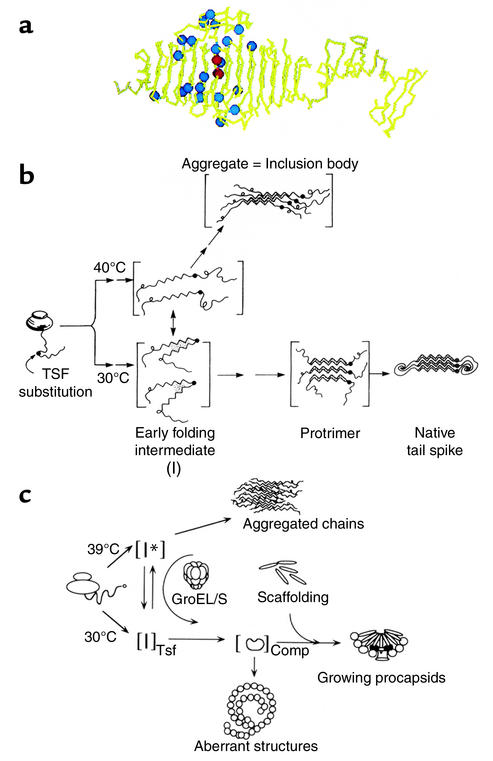
Protein misfolding and inclusion body formation studied in vivo with the phage P22 system. (a) Crystal structure of a subunit of the P22 tailspike homotrimer (20), showing the parallel β-coil structure and ventral and dorsal “fins.” Blue spheres represent positions of temperature-sensitive folding mutants (21), which in general occupy solvent-exposed sites in this native form (see text). Two red spheres represent two global suppressor mutations (23). Reproduced with permission from The FASEB Journal (22). (b) Pathways of folding and aggregation in vivo of the phage P22 tailspike protein bearing temperature-sensitive folding mutations. The single-chain folding intermediates are thermolabile (designated I), with the mutations, potentially affecting turn formation, favoring off-pathway intermediate formation and association over productive folding. The global suppressor mutations reverse such partitioning, favoring the productive early folding intermediate over the aggregation-producing one (vertical arrow), even at 40°C. Reproduced with permission from Science (23). TSF, temperature-sensitive folding. (c) Folding and aggregation in vivo of phage P22 capsid protein bearing temperature-sensitive folding mutations (24). Pathways are similar to those for the tailspike. Here, the chaperonin system, GroEL/GroES/ATP, can act at 39°C to bind the aggregation-prone I* intermediate (monomer) and facilitate its productive folding. Reproduced with permission from The Journal of Biological Chemistry (24).
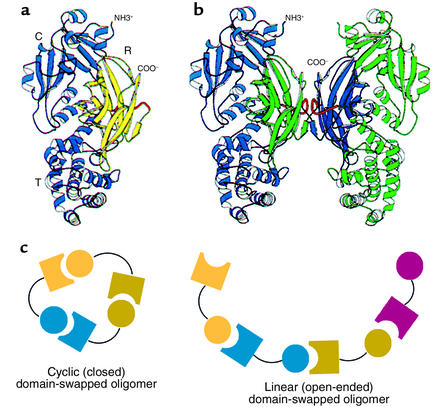
Domain swapping between protein monomers in crystals. (a and b) Original demonstration of domain swapping of diphtheria toxin in a protein crystal (26). The receptor-binding domain of a monomer (shown in yellow in a) can become rotated and translated to form contacts with the other two domains of the toxin, as in b, where the blue receptor-binding domain of the subunit at the left has become associated with the other two domains of the green subunit at the right, and vice versa. This demonstrates the substitution of interchain contacts for intrachain ones as proposed by Goldberg (see Figure 2). R, receptor binding domain. Reproduced with permission from Proceedings of the National Academy of Sciences of the United States of America (26). (c) Models for mechanics of higher-order domain swapping. Reproduced with permission from Protein Science (27).
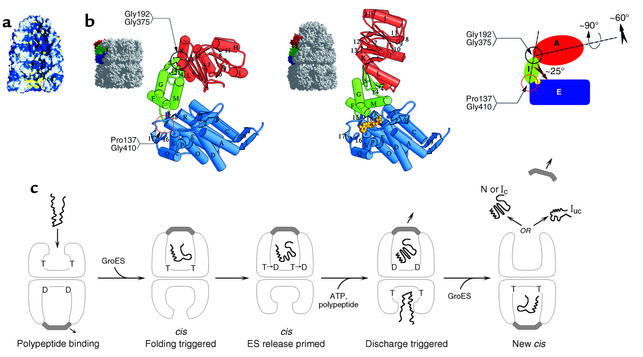
Chaperonin-mediated folding by GroEL-GroES. (a) Space-filling model of an asymmetric GroEL/GroES chaperonin complex showing GroES bound as a dome-shaped molecule to the upper GroEL ring with the lower GroEL ring open. The cavity of the lower ring displays a hydrophobic surface (yellow) that can accept non-native polypeptides exposing hydrophobic surface, while the cavity of the upper GroES-bound ring has displaced the hydrophobic surfaces away from the cavity, substituting them with hydrophilic surface (blue) that may serve to favor folding of the encapsulated polypeptide within this cavity. (b) Rigid body movements, associated with GroES binding to a GroEL ring, entail the elevation and twisting of the GroES-bound GroEL apical domains. This conformational change removes the hydrophobic surface from the cavity and replaces it with hydrophilic surface. Reproduced with permission from Nature (38). (c) Action of ATP binding and hydrolysis to advance the chaperonin cycle. ATP (designated as T; ADP as D) binds with positive cooperativity within one GroEL ring, but with negative cooperativity between rings, so that ATP effectively occupies only one ring at a time. This establishes the asymmetry of the system, which is reinforced by the nucleotide requirement for GroES binding. Non-native polypeptide binds to the open ring of an asymmetric complex (first panel), and GroES binding to the same ring forms a folding-active complex and triggers polypeptide folding (second panel). The lifetime of this complex is determined by ATP hydrolysis in the GroES-bound ring, which weakens the interaction between GroES and GroEL (third panel). Binding of ATP and polypeptide to the opposite ring (fourth panel) then discharges GroES and the polypeptide, either in the native state (N) or one committed to it (Ic) or in a still non-native state (Iuc) that can rebind to GroEL and try again to refold (fifth panel). Binding to an open GroEL ring may be associated with an unfolding action. Note that the rings oscillate back and forth as polypeptide-accepting and then folding-active, a function of the asymmetric binding of ATP/GroES. cis, binding of GroES and polypeptide to the same GroEL ring. Reproduced with permission from Current Opinion in Structural Biology (37).
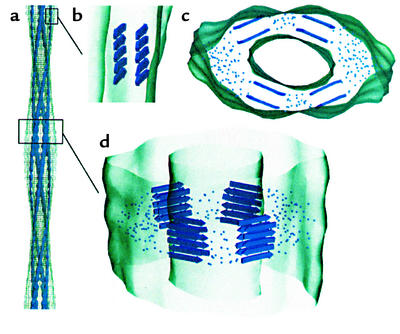
Model of an SH3 amyloid fibril as deduced from cryoEM microscopy of Saibil and coworkers (56). (a) Fibril structure showing four protofilaments and a fairly hollow interior space. (b) Side view of a single protofilament. (c and d) Model for packing of β-sheets into the fibril. Dots indicate regions of polypeptide connecting the β-sheet regions. Reproduced with permission from The EMBO Journal (56).
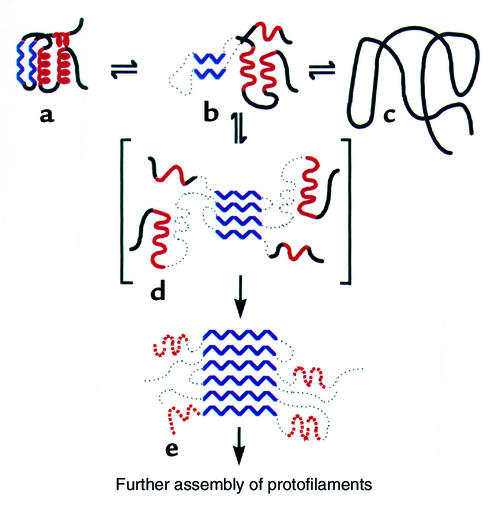
Mechanism proposed for lysozyme amyloidosis (59). An amyloidogenic intermediate state (b) formed from destabilization of the native structure (a) is proposed to self-associate through β-strand elements (blue) to initiate fibril formation (d, e), rather than forming non-native monomer (c). Reproduced with permission from Nature (59).
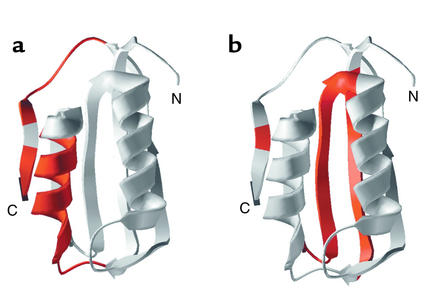
Distinct regions of acylphosphatase govern alternative fates of aggregation to form amyloid (a) or productive folding to native form (b). N, N-terminus; C, C-terminus of the polypepetide chain. Reproduced with permission from Nature Structural Biology (73).
References
-
- Weatherall, D.J., Clegg, J.B., Higgs, D.R., and Wood, W.G. 2001. The hemoglobinopathies. In Metabolic and molecular bases of inherited disease. C. Scriver, A. Beaudet, D. Valle, and W. Sly, editors. McGraw-Hill. New York, New York, USA. 4571–4636.
-
- Cox, D.W. 2001. α1-Antitrypsin deficiency. In Metabolic and molecular bases of inherited disease. C. Scriver, A. Beaudet, D. Valle, and W. Sly, editors. McGraw-Hill. New York, New York, USA. 5559–5584.
-
- Selkoe, D. 1997. Cellular and molecular biology of the beta-amyloid precursor protein and Alzheimer’s disease. In The molecular and genetic basis of neurological disease. R.N. Rosenberg, S.B. Prusiner, S. DiMauro, and R.L. Barchi, editors. Butterworth-Heinemann. Boston, Massachusetts, USA. 601–611.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
