

Failure to censor forbidden clones of CD4 T cells in autoimmune diabetes
- PMID: 12417628
- PMCID: PMC2194101
- DOI: 10.1084/jem.20020735
Failure to censor forbidden clones of CD4 T cells in autoimmune diabetes
Abstract
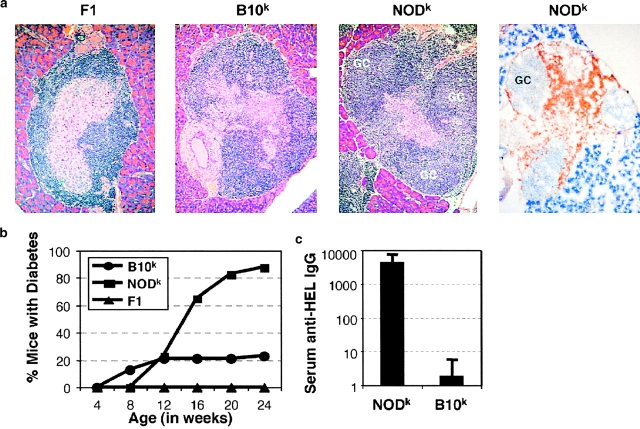
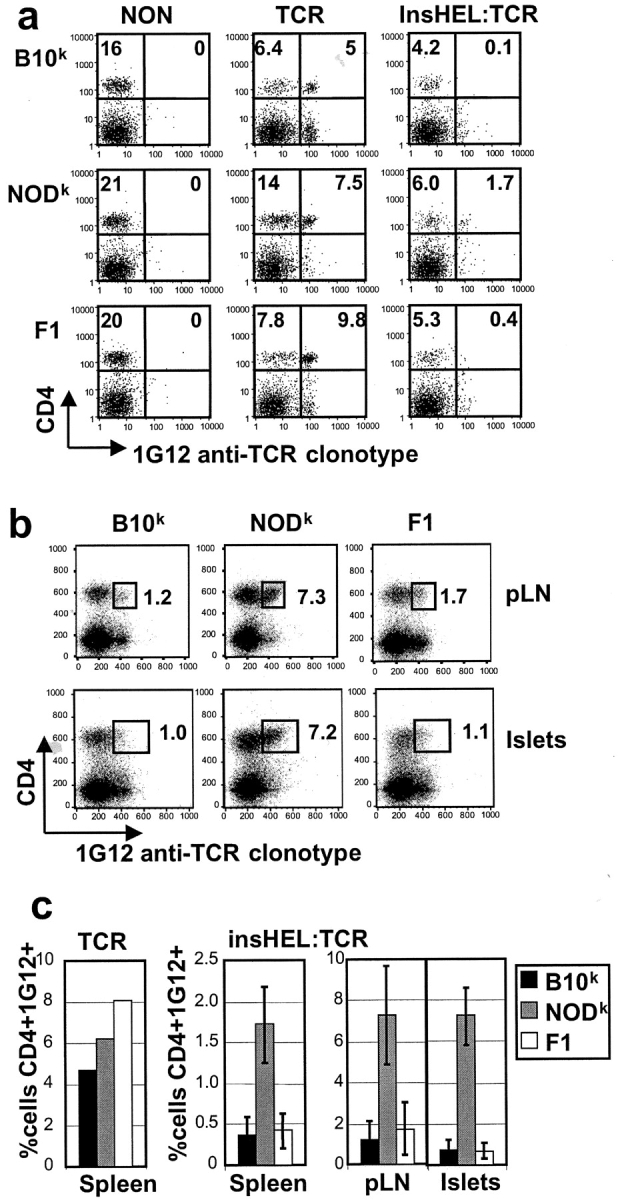
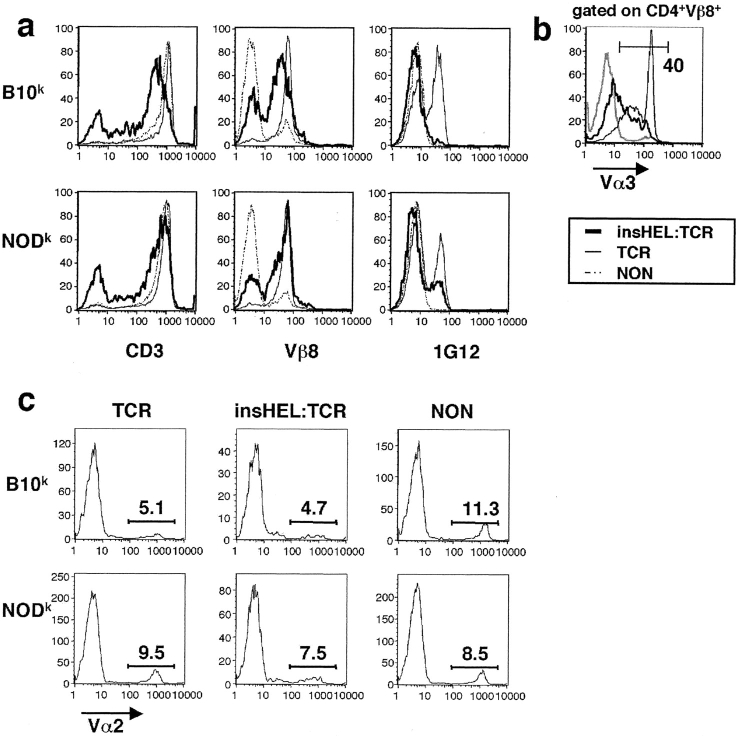
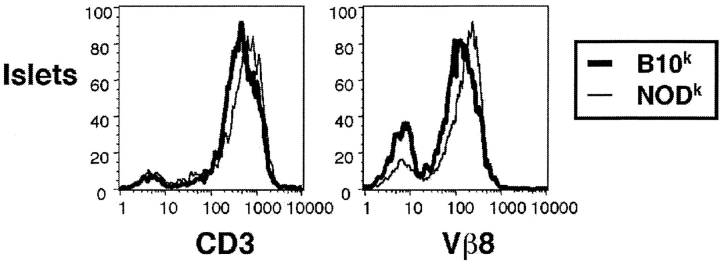
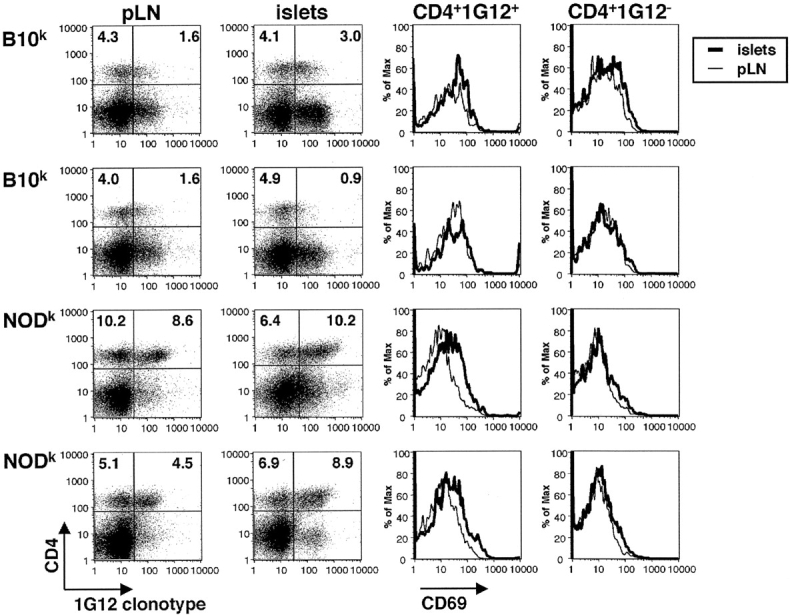
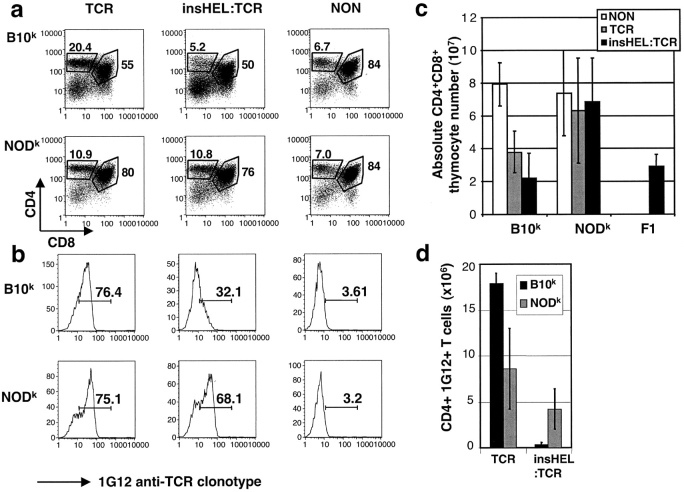
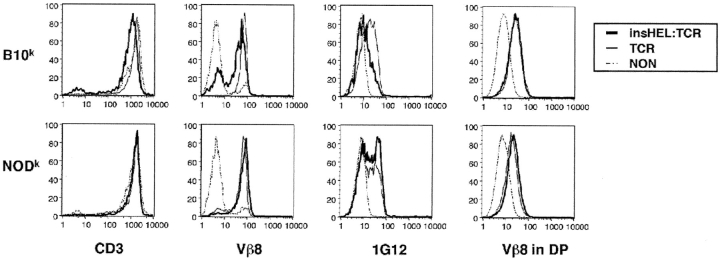
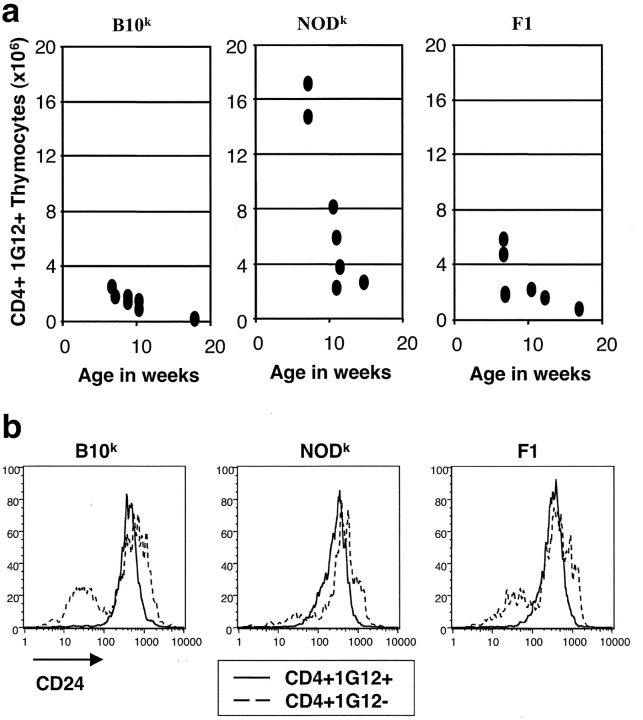
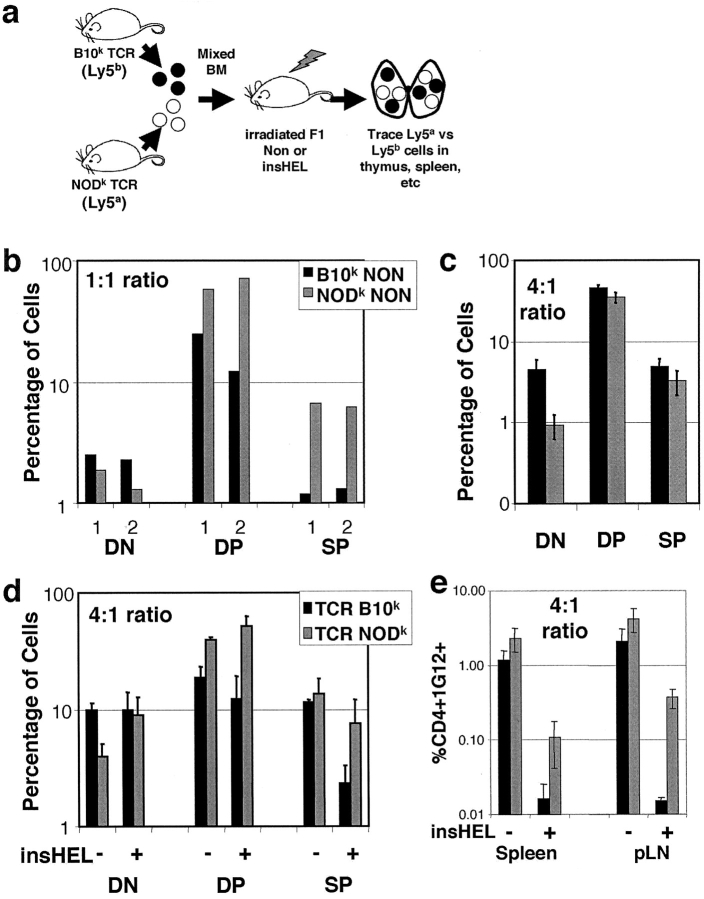
Type 1 diabetes and other organ-specific autoimmune diseases often cluster together in human families and in congenic strains of NOD (nonobese diabetic) mice, but the inherited immunoregulatory defects responsible for these diseases are unknown. Here we track the fate of high avidity CD4 T cells recognizing a self-antigen expressed in pancreatic islet beta cells using a transgenic mouse model. T cells of identical specificity, recognizing a dominant peptide from the same islet antigen and major histocompatibility complex (MHC)-presenting molecule, were followed on autoimmune susceptible and resistant genetic backgrounds. We show that non-MHC genes from the NOD strain cause a failure to delete these high avidity autoreactive T cells during their development in the thymus, with subsequent spontaneous breakdown of CD4 cell tolerance to the islet antigen, formation of intra-islet germinal centers, and high titre immunoglobulin G1 autoantibody production. In mixed bone marrow chimeric animals, defective thymic deletion was intrinsic to T cells carrying diabetes susceptibility genes. These results demonstrate a primary failure to censor forbidden clones of self-reactive T cells in inherited susceptibility to organ-specific autoimmune disease, and highlight the importance of thymic mechanisms of tolerance in organ-specific tolerance.
Figures
References
-
- Todd, J.A., and L.S. Wicker. 2001. Genetic protection from the inflammatory disease type 1 diabetes in humans and animal models. Immunity. 15:387–395. - PubMed
-
- Sinha, A.A., M.T. Lopez, and H.O. McDevitt. 1990. Autoimmune diseases: the failure of self tolerance. Science. 248:1380–1388. - PubMed
-
- Bach, J.F., and L. Chatenoud. 2001. Tolerance to islet autoantigens in Type 1 diabetes. Annu. Rev. Immunol. 19:131–161. - PubMed
-
- Adorini, L., S. Gregori, and L.C. Harrison. 2002. Understanding autoimmune diabetes: insights from mouse models. Trends Mol. Med. 8:31–38. - PubMed
-
- Delovitch, T.L., and B. Singh. 1997. The nonobese diabetic mouse as a model of autoimmune diabetes: immune dysregulation gets the NOD. Immunity. 7:727–738. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
