

Structural characterization of the human proteome
- PMID: 12421749
- PMCID: PMC187559
- DOI: 10.1101/gr.221202
Structural characterization of the human proteome
Abstract
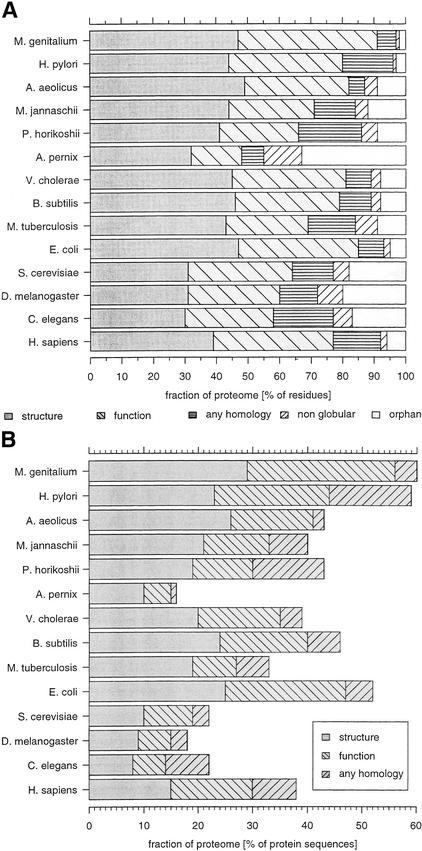
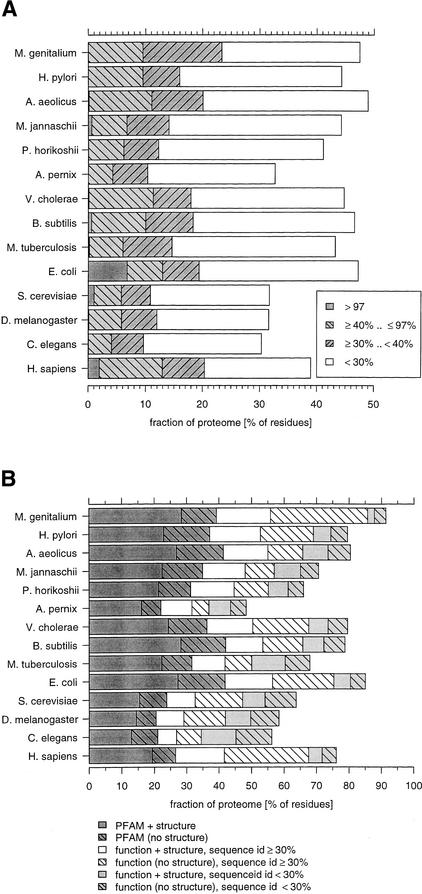
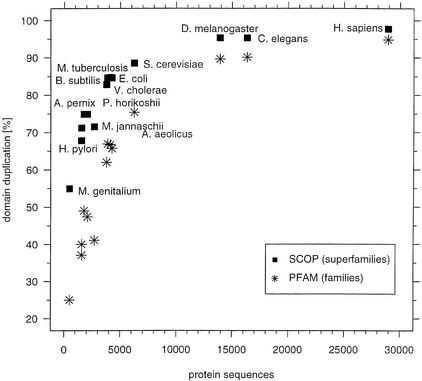
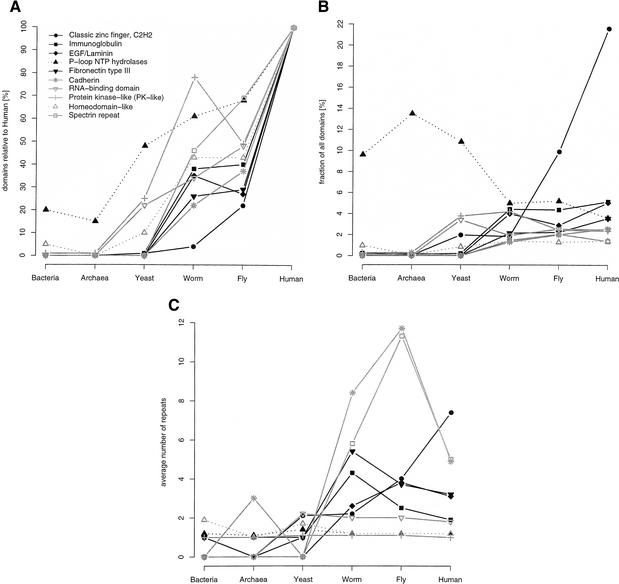
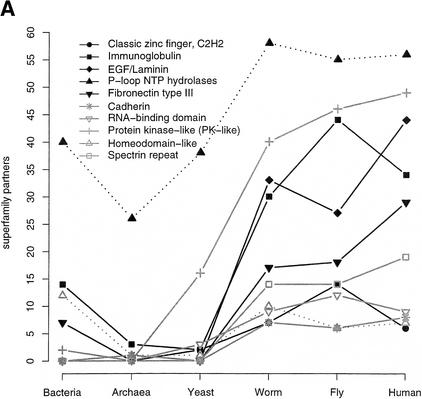
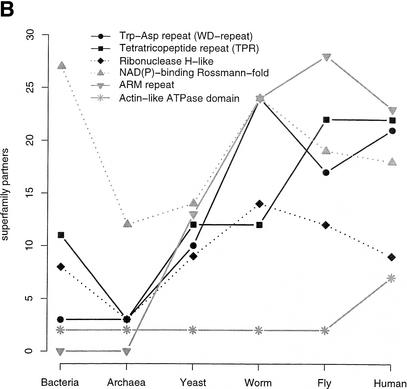
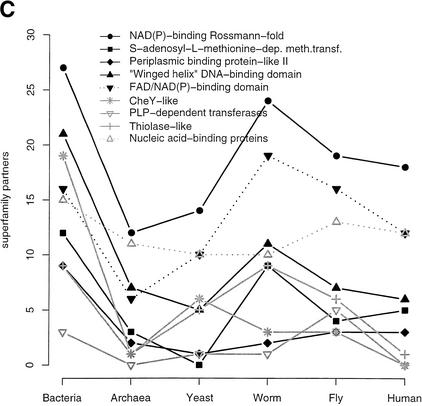
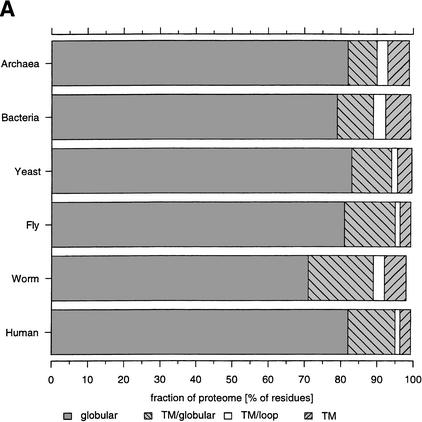
This paper reports an analysis of the encoded proteins (the proteome) of the genomes of human, fly, worm, yeast, and representatives of bacteria and archaea in terms of the three-dimensional structures of their globular domains together with a general sequence-based study. We show that 39% of the human proteome can be assigned to known structures. We estimate that for 77% of the proteome, there is some functional annotation, but only 26% of the proteome can be assigned to standard sequence motifs that characterize function. Of the human protein sequences, 13% are transmembrane proteins, but only 3% of the residues in the proteome form membrane-spanning regions. There are substantial differences in the composition of globular domains of transmembrane proteins between the proteomes we have analyzed. Commonly occurring structural superfamilies are identified within the proteome. The frequencies of these superfamilies enable us to estimate that 98% of the human proteome evolved by domain duplication, with four of the 10 most duplicated superfamilies specific for multicellular organisms. The zinc-finger superfamily is massively duplicated in human compared to fly and worm, and occurrence of domains in repeats is more common in metazoa than in single cellular organisms. Structural superfamilies over- and underrepresented in human disease genes have been identified. Data and results can be downloaded and analyzed via web-based applications at http://www.sbg.bio.ic.ac.uk.
Figures
Similar articles
-
Molecular phylogeny of the kelch-repeat superfamily reveals an expansion of BTB/kelch proteins in animals.BMC Bioinformatics. 2003 Sep 17;4:42. doi: 10.1186/1471-2105-4-42. Epub 2003 Sep 17. BMC Bioinformatics. 2003. PMID: 13678422 Free PMC article.
-
Multi-modality of pI distribution in whole proteome.Proteomics. 2006 Jan;6(2):449-55. doi: 10.1002/pmic.200500221. Proteomics. 2006. PMID: 16317776
-
Comparative analysis of protein domain organization.Genome Res. 2004 Mar;14(3):343-53. doi: 10.1101/gr.1610504. Genome Res. 2004. PMID: 14993202 Free PMC article.
-
[Proteins sharing PNPLA domain, a new family of enzymes regulating lipid metabolism].Med Sci (Paris). 2010 Feb;26(2):177-84. doi: 10.1051/medsci/2010262177. Med Sci (Paris). 2010. PMID: 20188050 Review. French.
-
The proteome: structure, function and evolution.Philos Trans R Soc Lond B Biol Sci. 2006 Mar 29;361(1467):441-51. doi: 10.1098/rstb.2005.1802. Philos Trans R Soc Lond B Biol Sci. 2006. PMID: 16524832 Free PMC article. Review.
Cited by
-
Structural characterization of genomes by large scale sequence-structure threading.BMC Bioinformatics. 2004 Apr 3;5:37. doi: 10.1186/1471-2105-5-37. BMC Bioinformatics. 2004. PMID: 15061866 Free PMC article.
-
prot4EST: translating expressed sequence tags from neglected genomes.BMC Bioinformatics. 2004 Nov 30;5:187. doi: 10.1186/1471-2105-5-187. BMC Bioinformatics. 2004. PMID: 15571632 Free PMC article.
-
Global patterns of protein domain gain and loss in superkingdoms.PLoS Comput Biol. 2014 Jan 30;10(1):e1003452. doi: 10.1371/journal.pcbi.1003452. eCollection 2014 Jan. PLoS Comput Biol. 2014. PMID: 24499935 Free PMC article.
-
Intramolecular interaction in the tail of Acanthamoeba myosin IC between the SH3 domain and a putative pleckstrin homology domain.Proc Natl Acad Sci U S A. 2007 Jan 16;104(3):784-9. doi: 10.1073/pnas.0610231104. Epub 2007 Jan 10. Proc Natl Acad Sci U S A. 2007. PMID: 17215368 Free PMC article.
-
Phylogeny of Toll-like receptor signaling: adapting the innate response.PLoS One. 2013;8(1):e54156. doi: 10.1371/journal.pone.0054156. Epub 2013 Jan 11. PLoS One. 2013. PMID: 23326591 Free PMC article.
References
-
- Aloy P, Querol E, Aviles FX, Sternberg MJE. Automated structure-based prediction of functional sites in proteins—Applications to assessing the validity of inheriting protein function from homology in genome annotation and to protein docking. J Mol Biol. 2001;311:395–408. - PubMed
-
- Antonarakis SE, McKusick VA. OMIM passes the 1,000-disease-gene mark. Nat Genet. 2000;25:11. - PubMed
-
- Apic G, Gough J, Teichmann SA. Domain combinations inarchaeal, eubacterial and eukaryotic proteomes. J Mol Biol. 2001;310:311–325. - PubMed
-
- Bargmann CI. Neurobiology of the Caenorhabditis elegans genome. Science. 1998;282:2028–2033. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases