

Structural and functional characterization of factor H mutations associated with atypical hemolytic uremic syndrome
- PMID: 12424708
- PMCID: PMC378565
- DOI: 10.1086/344515
Structural and functional characterization of factor H mutations associated with atypical hemolytic uremic syndrome
Abstract
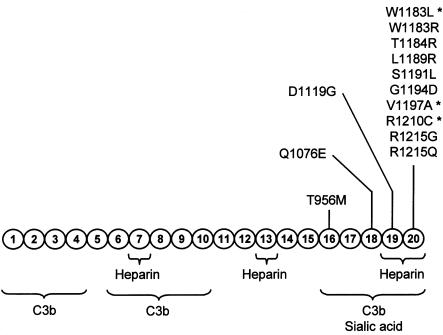
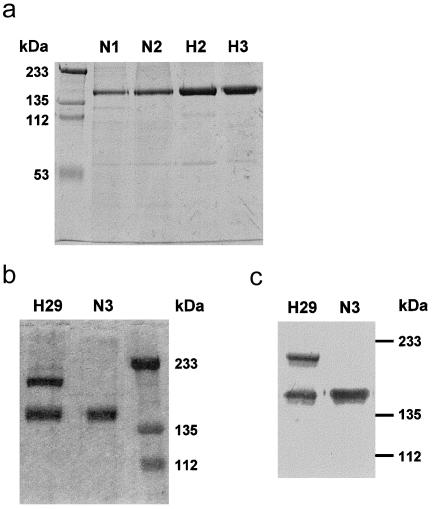
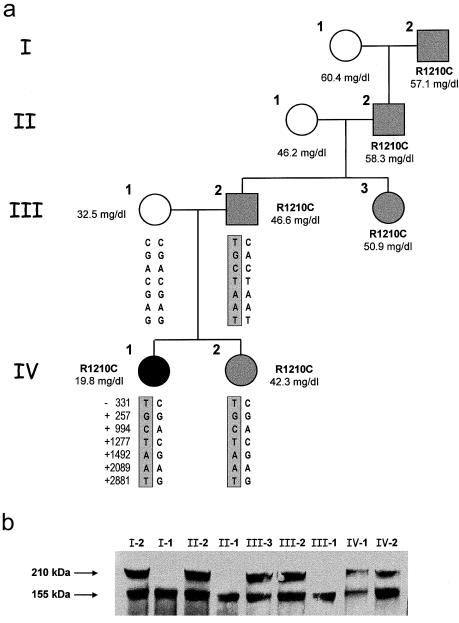
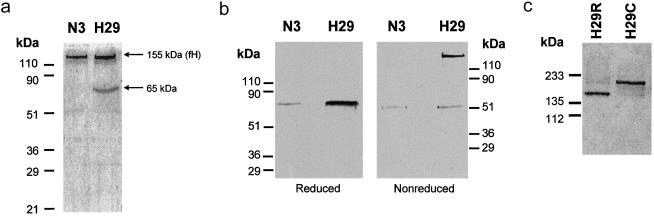
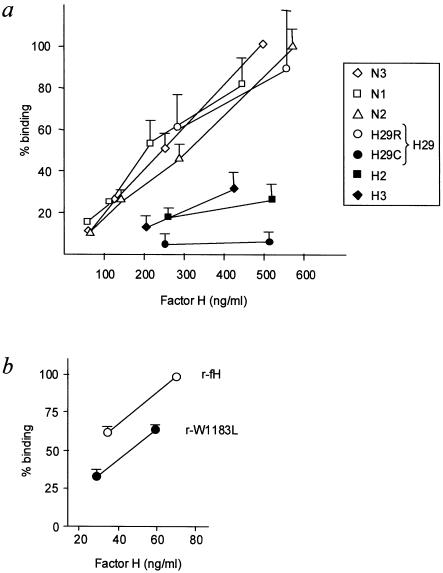
Genetic studies have demonstrated the involvement of the complement regulator factor H in nondiarrheal, nonverocytotoxin (i.e., atypical) cases of hemolytic uremic syndrome. Different factor H mutations have been identified in 10%-30% of patients with atypical hemolytic uremic syndrome (aHUS), and most of these mutations alter single amino acids in the C-terminal region of factor H. Although these mutations are considered to be responsible for the disease, the precise role that factor H plays in the pathogenesis of aHUS is unknown. We report here the structural and functional characterization of three different factor H proteins purified from the plasma of patients with aHUS who carry the factor H mutations W1183L, V1197A, or R1210C. Structural anomalies in factor H were found only in R1210C carriers; these individuals show, in their plasma, a characteristic high-molecular-weight factor H protein that results from the covalent interaction between factor H and human serum albumin. Most important, all three aHUS-associated factor H proteins have a normal cofactor activity in the proteolysis of fluid-phase C3b by factor I but show very low binding to surface-bound C3b. This functional impairment was also demonstrated in recombinant mutant factor H proteins expressed in COS7 cells. These data support the hypothesis that patients with aHUS carry a specific dysfunction in the protection of cellular surfaces from complement activation, offering new possibilities to improve diagnosis and develop appropriate therapies.
Figures

References
Electronic-Database Information
-
- Entrez-Nucleotide, http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=Nucleotide (for factor H cDNA sequence [accession number Y00716])
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for autosomal dominant [MIM 134370] and recessive [MIM 235400] HUS)
References
-
- Blackmore TK, Hellwage J, Sadlon TA, Higgs N, Zipfel PF, Ward HM, Gordon DL (1998) Identification of the second heparin-binding domain in human complement factor H. J Immunol 160:3342–3348 - PubMed
-
- Blackmore TK, Sadlon TA, Ward HM, Lublin DM, Gordon DL (1996) Identification of a heparin binding domain in the seventh short consensus repeat of complement factor H. J Immunol 157:5422–5427 - PubMed
-
- Caprioli J, Bettinaglio P, Zipfel PF, Amadei B, Daina E, Gamba S, Skerka C, Marziliano N, Remuzzi G, Noris M (2001) The molecular basis of familial hemolytic uremic syndrome: mutation analysis of factor H gene reveals a hot spot in short consensus repeat 20. J Am Soc Nephrol 12:297–307 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
