

Review
doi: 10.1136/heart.88.6.658.
Pulmonary hypertension in the young
Affiliations
- PMID: 12433912
- PMCID: PMC1767475
- DOI: 10.1136/heart.88.6.658
Item in Clipboard
Review
Pulmonary hypertension in the young
Heart.
2002 Dec.
No abstract available
Figures
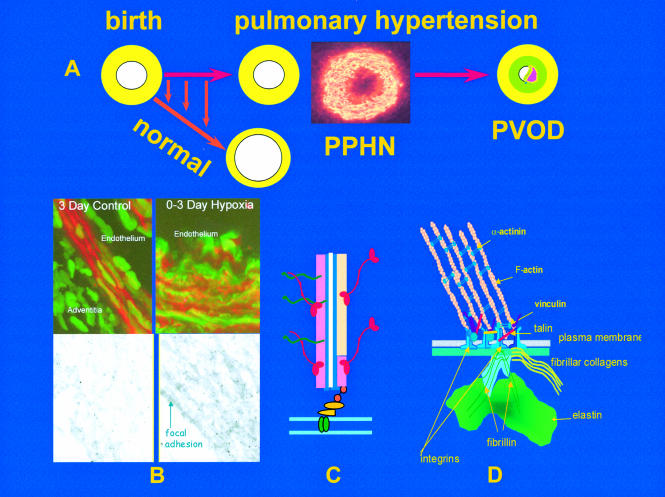
The upper figure (A) illustrates the rapid reduction in pulmonary arterial wall thickness occurring immediately after birth in the normal lung. This process is profoundly disturbed in persistent pulmonary hypertension of the newborn (PPHN) and an increase in medial thickness eventually leads to pulmonary vascular obstructive disease (PVOD) if the pressure remains high. Insert shows abnormal, hypertensive human peripheral pulmonary artery at three days, stained for γ actin. Mechanisms are illustrated in B, C, and D. (B) Confocal and transmission electron microscopy shows, in the left hand panel, the normal porcine peripheral pulmonary artery, and in the right hand panel, the pulmonary hypertensive vessel at three days. Normal remodelling entails reorganisation of the smooth muscle cell actin cytoskeleton which undergoes transient disassembly as the cells thin and elongate to spread around an enlarging lumen. In PPHN larger cells are packed with red actin myofilaments (phalloidin stained) while immuno-electron microcopy reveals sheets of actin bundles labelled with gold particles rather than actin being diffusely distributed in the cytosol. (C) Illustration of a myofilament. (D) All aspects of vessel wall remodelling are disturbed in PPHN, from the excessive myofilament assembly within the cell, focal adhesion remodelling at the membrane, and excessive connective tissue deposition in the matrix. Gene expression of tropoelastin and type I procollagen is abnormally high and steady state protein concentrations are increased. Vessels appear to become fixed in an incompletely dilated state. This is the beginning of PVOD in the young if the process cannot be arrested therapeutically.
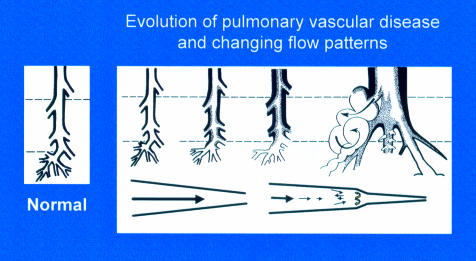
Evolution of pulmonary vascular disease in the young showing the early reduction in number of arteries, the increase in muscularity, and development of intimal proliferation. Two classification systems are used to describe these changes, the more recent concentrating on the early structural changes preceding intimal damage in a pulmonary vascular bed which is still developing, the other being the Heath and Edwards classification. A biopsy must be taken at sufficient depth (upper interrupted line) to include some pre-acinar arteries in which intimal proliferation first develops. For a limited time the more peripheral arteries can have a misleading near-normal appearance, a predilatation phase associated with an increase in mortality and morbidity at and after intracardiac repair. Blood flow patterns change from laminar to turbulent with disease progression.
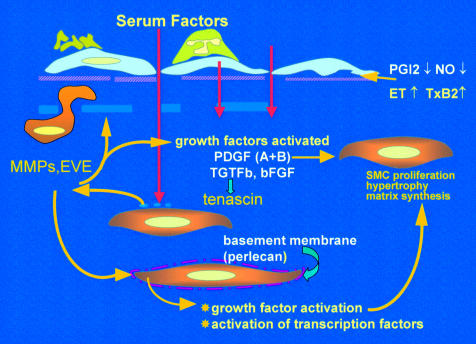
Factors driving the evolution of pulmonary vascular obstructive disease. Endothelial dysfunction reduces release of prostacyclin and nitric oxide (NO), causes adherence of activated platelets and leucocytes, enhanced release of thromboxane and endothelin, and loss of barrier function with leakage of serum factor into the subendothelium. This last action is thought to heighten activity of metalloproteinases (MMPs), including the proteolytic enzyme endogenous vascular elastase (EVE) released from smooth muscle cells, to help induce structural remodelling, cause smooth muscle cell activation, disruption of the internal elastic lamina, and facilitate smooth muscle cell migration. MMPs also activate growth factors normally sequestered in the matrix in an inactive form. Increased tenascin expression is associated with cell proliferation, its downregulation with apoptosis. Tenascin amplifies the proliferative response to epidermal growth factor and fibroblast growth factor (FGF)-2 in vitro. Expression of fibronectin is widespread and this glycoprotein can facilitate smooth muscle migration. The innermost smooth muscle cells cease to express many smooth muscle specific contractile and cytoskeletal proteins before migrating through gaps in the internal elastic lamina. Changes in phenotype are widespread.
References
-
- Rich S. Primary pulmonary hypertension: executive summary from the world symposium on primary pulmonary hypertension 1998, Evian, France, 6–10 September 1998. Rich S, 2002. ▸ Recent WHO consensus classification of pulmonary hypertension.
-
- Haworth SG. Pathobiology of pulmonary hypertension in infants and children. Prog Ped Card 2001;12:249–69. ▸ Useful review.
-
- Tulloh RMR, Hislop AA, Boels PJ, et al. Chronic hypoxia inhibits postnatal maturation of porcine intrapulmonary artery relaxation. Am J Physiol 1997;272:H2436–45. - PubMed
-
- The International PPH Consortium, Lane KB, Machado RD, Pauciulo MW, et al. Heterozygous germline mutations in a TGF-β receptor, BMPR2, are the cause of familial primary pulmonary hypertension. Genetics 2000;26:81–4. ▸ The first paper reporting germline mutations in the BMPR2 gene in FPPH. - PubMed
-
- Johnson DW, Berg JN, Baldwin MA, et al. Mutations in the activin receptor-like kinase 1 gene in hereditary haemorrhagic telangiectasia type 2. Nat Genet 1996;13:189–95. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical