

3-Methylglutaconic aciduria type I is caused by mutations in AUH
- PMID: 12434311
- PMCID: PMC378594
- DOI: 10.1086/344712
3-Methylglutaconic aciduria type I is caused by mutations in AUH
Erratum in
- Am J Hum Genet. 2003 Sep;73(3):709
Abstract
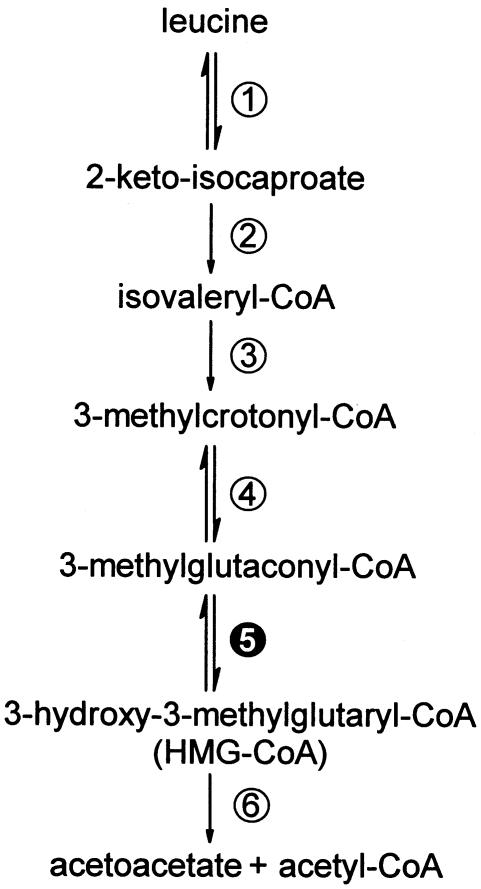
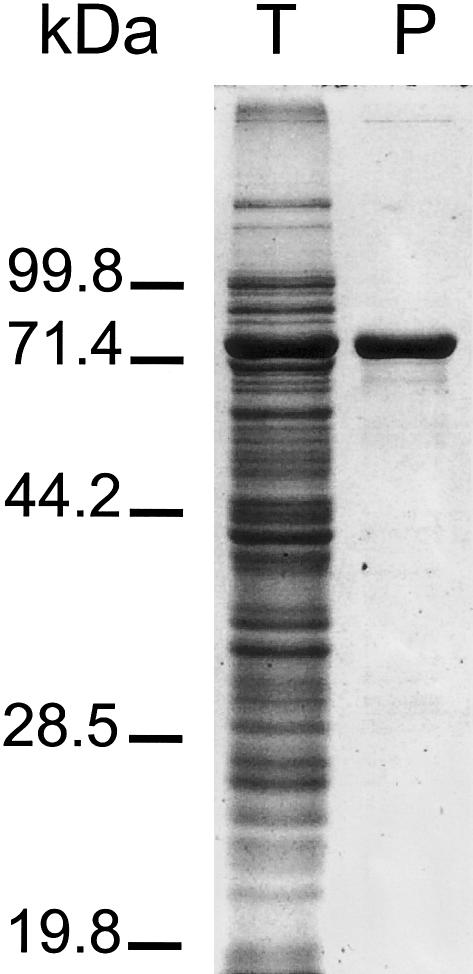
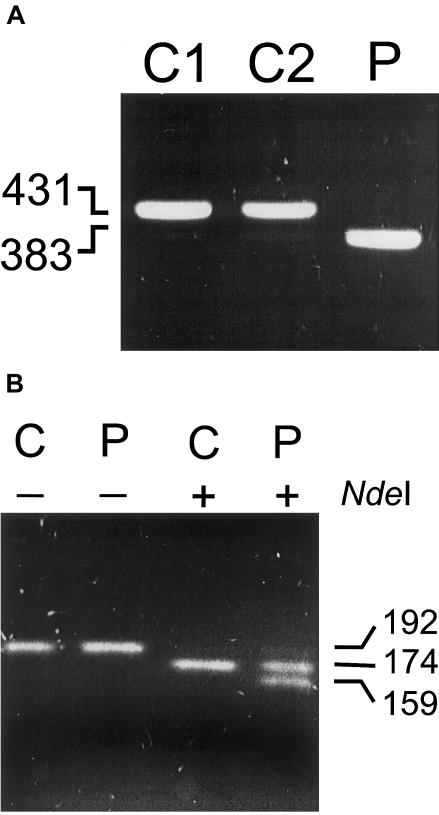
3-Methylglutaconic aciduria type I is an autosomal recessive disorder clinically characterized by various symptoms ranging from delayed speech development to severe neurological handicap. This disorder is caused by a deficiency of 3-methylglutaconyl-CoA hydratase, one of the key enzymes of leucine degradation. This results in elevated urinary levels of 3-methylglutaconic acid, 3-methylglutaric acid, and 3-hydroxyisovaleric acid. By heterologous expression in Escherichia coli, we show that 3-methylglutaconyl-CoA hydratase is encoded by the AUH gene, whose product had been reported elsewhere as an AU-specific RNA-binding protein. Mutation analysis of AUH in two patients revealed a nonsense mutation (R197X) and a splice-site mutation (IVS8-1G-->A), demonstrating that mutations in AUH cause 3-methylglutaconic aciduria type I.
Figures
Similar articles
-
Mutations in the AUH gene cause 3-methylglutaconic aciduria type I.Hum Mutat. 2003 Apr;21(4):401-7. doi: 10.1002/humu.10202. Hum Mutat. 2003. PMID: 12655555
-
Biochemical characterization of human 3-methylglutaconyl-CoA hydratase and its role in leucine metabolism.FEBS J. 2006 May;273(9):2012-22. doi: 10.1111/j.1742-4658.2006.05218.x. FEBS J. 2006. PMID: 16640564
-
3-methylglutaconic aciduria type I in a boy with fever-associated seizures.Pediatr Neurol. 2004 Mar;30(3):213-5. doi: 10.1016/j.pediatrneurol.2003.09.016. Pediatr Neurol. 2004. PMID: 15033206
-
Early infantile presentation of 3-methylglutaconic aciduria type 1 with a novel mutation in AUH gene: A case report and literature review.Brain Dev. 2017 Sep;39(8):714-716. doi: 10.1016/j.braindev.2017.04.007. Epub 2017 Apr 21. Brain Dev. 2017. PMID: 28438368 Review.
-
3-Methylglutaconic Aciduria Type I Due to AUH Defect: The Case Report of a Diagnostic Odyssey and a Review of the Literature.Int J Mol Sci. 2022 Apr 16;23(8):4422. doi: 10.3390/ijms23084422. Int J Mol Sci. 2022. PMID: 35457240 Free PMC article. Review.
Cited by
-
Exposure to environmental radionuclides is associated with altered metabolic and immunity pathways in a wild rodent.Mol Ecol. 2019 Oct;28(20):4620-4635. doi: 10.1111/mec.15241. Epub 2019 Sep 30. Mol Ecol. 2019. PMID: 31498518 Free PMC article.
-
Mitochondrial dysfunction and organic aciduria in five patients carrying mutations in the Ras-MAPK pathway.Eur J Hum Genet. 2011 Feb;19(2):138-44. doi: 10.1038/ejhg.2010.171. Epub 2010 Nov 10. Eur J Hum Genet. 2011. PMID: 21063443 Free PMC article.
-
Overcoming the divide between ataxias and spastic paraplegias: Shared phenotypes, genes, and pathways.Mov Disord. 2017 Mar;32(3):332-345. doi: 10.1002/mds.26944. Epub 2017 Feb 14. Mov Disord. 2017. PMID: 28195350 Free PMC article. Review.
-
Metabolic and regulatory roles of leucine in neural cells.Neurochem Res. 2008 Feb;33(2):279-84. doi: 10.1007/s11064-007-9444-4. Epub 2007 Aug 25. Neurochem Res. 2008. PMID: 17721727 Review.
-
OPA3, mutated in 3-methylglutaconic aciduria type III, encodes two transcripts targeted primarily to mitochondria.Mol Genet Metab. 2010 Jun;100(2):149-54. doi: 10.1016/j.ymgme.2010.03.005. Epub 2010 Mar 16. Mol Genet Metab. 2010. PMID: 20350831 Free PMC article.
References
Electronic-Database Information
-
- GenBank, http://www.ncbi.nlm.nih.gov/Genbank/ (for AUH [accession numbers NM_001698 and NT_008476]
References
-
- Duran M, Beemer FA, Tibosch AS, Bruinvis L, Ketting D, Wadman SK (1982) Inherited 3-methylglutaconic aciduria in two brothers: another defect of leucine metabolism. J Pediatr 101:551–554 - PubMed
-
- Ensenauer R, Muller CB, Schwab KO, Gibson KM, Brandis M, Lehnert W (2000) 3-Methylglutaconyl-CoA hydratase deficiency: a new patient with speech retardation as the leading sign. J Inherit Metab Dis 23:341–344 - PubMed
-
- Gibson KM, Lee CF, Wappner RS (1992) 3-Methylglutaconyl-coenzyme—a hydratase deficiency: a new case. J Inherit Metab Dis 15:363–366 - PubMed
-
- Gibson KM, Wappner RS, Jooste S, Erasmus E, Mienie LJ, Gerlo E, Desprechins B, De Meirleir L (1998) Variable clinical presentation in three patients with 3-methylglutaconyl-coenzyme A hydratase deficiency. J Inherit Metab Dis 21:631–638 - PubMed
-
- Hilz H, Knappe J, Ringelmann E, Lynen F (1958) Methylglutaconase, eine neue hydratase, die am stoffwechsel verzweigter carbonsäuren beteiligt ist. Biochem Z 329:476–489 - PubMed
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
