

Transmembrane helix predictions revisited
- PMID: 12441377
- PMCID: PMC2373751
- DOI: 10.1110/ps.0214502
Transmembrane helix predictions revisited
Abstract
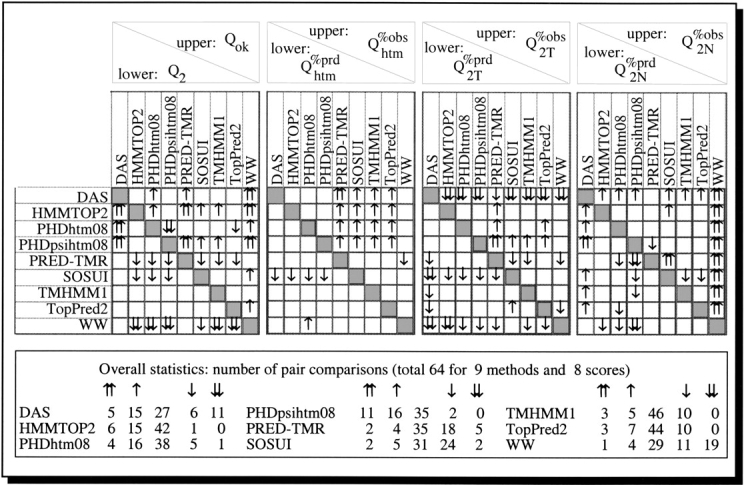
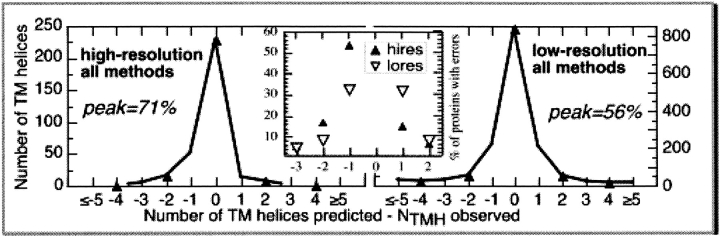
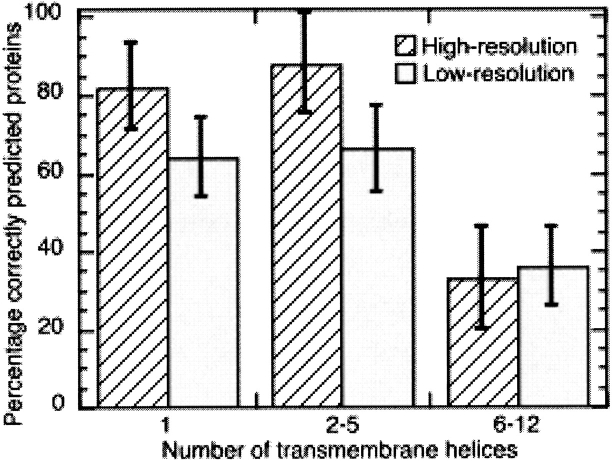
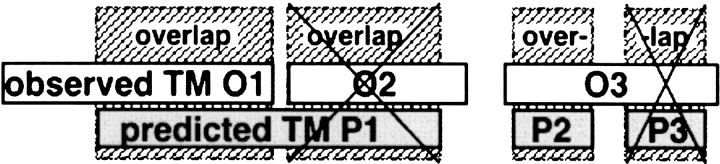
Methods that predict membrane helices have become increasingly useful in the context of analyzing entire proteomes, as well as in everyday sequence analysis. Here, we analyzed 27 advanced and simple methods in detail. To resolve contradictions in previous works and to reevaluate transmembrane helix prediction algorithms, we introduced an analysis that distinguished between performance on redundancy-reduced high- and low-resolution data sets, established thresholds for significant differences in performance, and implemented both per-segment and per-residue analysis of membrane helix predictions. Although some of the advanced methods performed better than others, we showed in a thorough bootstrapping experiment based on various measures of accuracy that no method performed consistently best. In contrast, most simple hydrophobicity scale-based methods were significantly less accurate than any advanced method as they overpredicted membrane helices and confused membrane helices with hydrophobic regions outside of membranes. In contrast, the advanced methods usually distinguished correctly between membrane-helical and other proteins. Nonetheless, few methods reliably distinguished between signal peptides and membrane helices. We could not verify a significant difference in performance between eukaryotic and prokaryotic proteins. Surprisingly, we found that proteins with more than five helices were predicted at a significantly lower accuracy than proteins with five or fewer. The important implication is that structurally unsolved multispanning membrane proteins, which are often important drug targets, will remain problematic for transmembrane helix prediction algorithms. Overall, by establishing a standardized methodology for transmembrane helix prediction evaluation, we have resolved differences among previous works and presented novel trends that may impact the analysis of entire proteomes.
Figures

References
-
- Altschul, S.F. and Gish, W. 1996. Local alignment statistics. Meth. Enzymol. 266 460–480. - PubMed
-
- Amstutz, P., Forrer, P., Zahnd, C., and Pluckthun, A. 2001. In vitro display technologies: Novel developments and applications. Curr. Opin. Biotechnol. 12 400–405. - PubMed
-
- Bauer, M.F., Hofmann, S., Neupert, W., and Brunner, M. 2000. Protein translocation into mitochondria: The role of TIM complexes. TICB 10 25–31. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
