

Thoroughly sampling sequence space: large-scale protein design of structural ensembles
- PMID: 12441379
- PMCID: PMC2373757
- DOI: 10.1110/ps.0203902
Thoroughly sampling sequence space: large-scale protein design of structural ensembles
Abstract
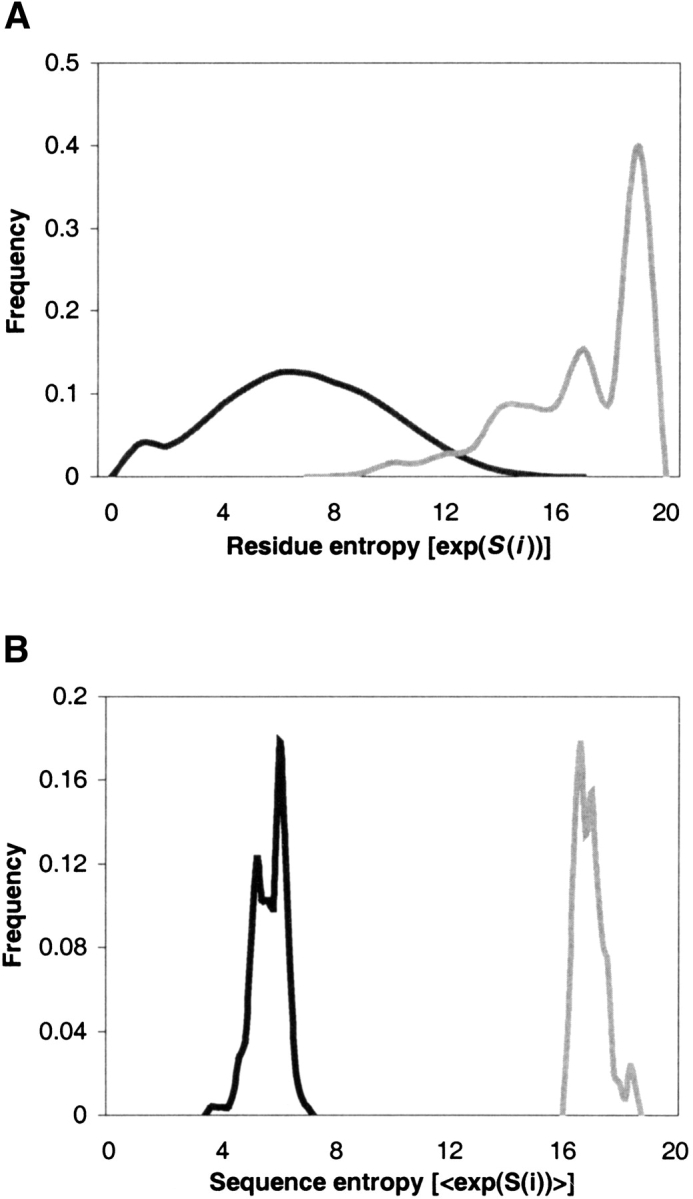
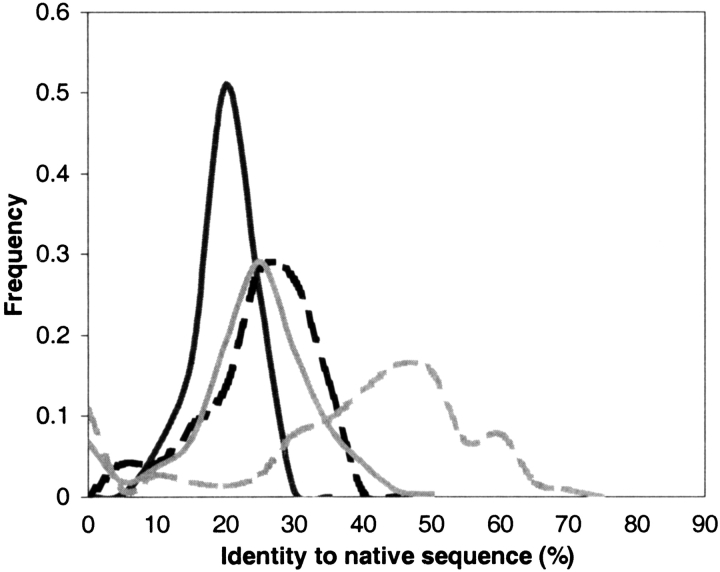
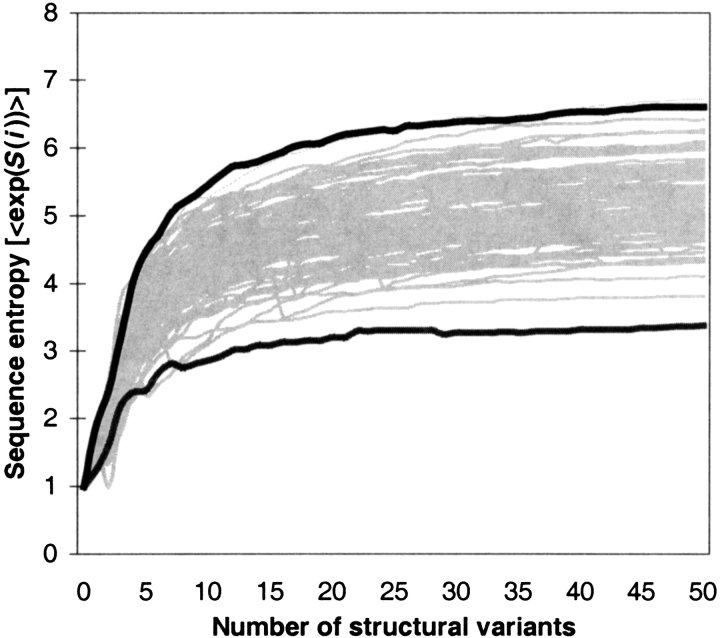
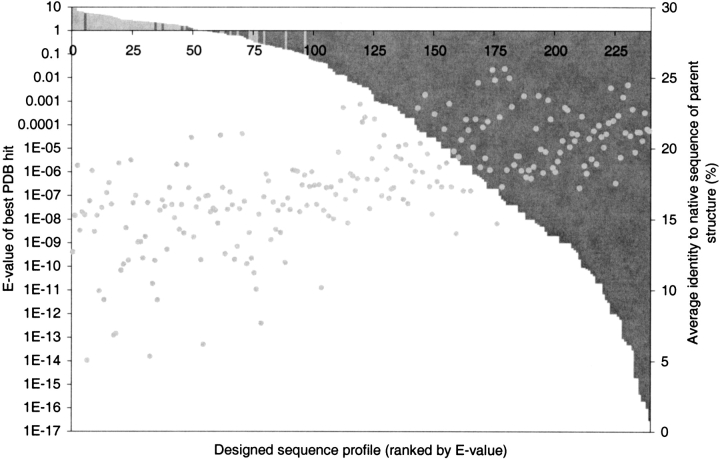
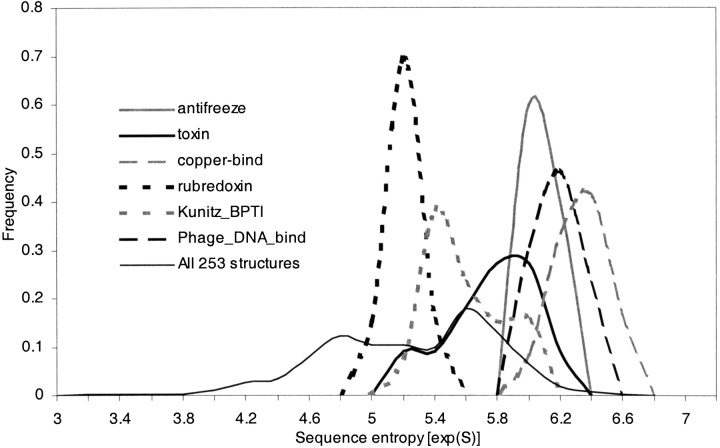
Modeling the inherent flexibility of the protein backbone as part of computational protein design is necessary to capture the behavior of real proteins and is a prerequisite for the accurate exploration of protein sequence space. We present the results of a broad exploration of sequence space, with backbone flexibility, through a novel approach: large-scale protein design to structural ensembles. A distributed computing architecture has allowed us to generate hundreds of thousands of diverse sequences for a set of 253 naturally occurring proteins, allowing exciting insights into the nature of protein sequence space. Designing to a structural ensemble produces a much greater diversity of sequences than previous studies have reported, and homology searches using profiles derived from the designed sequences against the Protein Data Bank show that the relevance and quality of the sequences is not diminished. The designed sequences have greater overall diversity than corresponding natural sequence alignments, and no direct correlations are seen between the diversity of natural sequence alignments and the diversity of the corresponding designed sequences. For structures in the same fold, the sequence entropies of the designed sequences cluster together tightly. This tight clustering of sequence entropies within a fold and the separation of sequence entropy distributions for different folds suggest that the diversity of designed sequences is primarily determined by a structure's overall fold, and that the designability principle postulated from studies of simple models holds in real proteins. This has important implications for experimental protein design and engineering, as well as providing insight into protein evolution.
Figures
Similar articles
-
Accurate prediction for atomic-level protein design and its application in diversifying the near-optimal sequence space.Proteins. 2009 May 15;75(3):682-705. doi: 10.1002/prot.22280. Proteins. 2009. PMID: 19003998
-
Computational protein design: validation and possible relevance as a tool for homology searching and fold recognition.PLoS One. 2010 May 5;5(5):e10410. doi: 10.1371/journal.pone.0010410. PLoS One. 2010. PMID: 20463972 Free PMC article.
-
Filling-in void and sparse regions in protein sequence space by protein-like artificial sequences enables remarkable enhancement in remote homology detection capability.J Mol Biol. 2014 Feb 20;426(4):962-79. doi: 10.1016/j.jmb.2013.11.026. Epub 2013 Dec 4. J Mol Biol. 2014. PMID: 24316367
-
Computational protein design with backbone plasticity.Biochem Soc Trans. 2016 Oct 15;44(5):1523-1529. doi: 10.1042/BST20160155. Epub 2016 Oct 19. Biochem Soc Trans. 2016. PMID: 27911735 Free PMC article. Review.
-
Computational protein design: a novel path to future protein drugs.Curr Pharm Des. 2006;12(31):3973-97. doi: 10.2174/138161206778743655. Curr Pharm Des. 2006. PMID: 17100608 Review.
Cited by
-
Computationally designed libraries of fluorescent proteins evaluated by preservation and diversity of function.Proc Natl Acad Sci U S A. 2007 Jan 2;104(1):48-53. doi: 10.1073/pnas.0609647103. Epub 2006 Dec 19. Proc Natl Acad Sci U S A. 2007. PMID: 17179210 Free PMC article.
-
Toward full-sequence de novo protein design with flexible templates for human beta-defensin-2.Biophys J. 2008 Jan 15;94(2):584-99. doi: 10.1529/biophysj.107.110627. Epub 2007 Sep 7. Biophys J. 2008. PMID: 17827237 Free PMC article.
-
Predicting the tolerated sequences for proteins and protein interfaces using RosettaBackrub flexible backbone design.PLoS One. 2011;6(7):e20451. doi: 10.1371/journal.pone.0020451. Epub 2011 Jul 18. PLoS One. 2011. PMID: 21789164 Free PMC article.
-
Semirational Directed Evolution of Loop Regions in Aspergillus japonicus β-Fructofuranosidase for Improved Fructooligosaccharide Production.Appl Environ Microbiol. 2015 Oct;81(20):7319-29. doi: 10.1128/AEM.02134-15. Epub 2015 Aug 7. Appl Environ Microbiol. 2015. PMID: 26253664 Free PMC article.
-
Use of designed sequences in protein structure recognition.Biol Direct. 2018 May 9;13(1):8. doi: 10.1186/s13062-018-0209-6. Biol Direct. 2018. PMID: 29776380 Free PMC article.
References
-
- Baldwin, E.P., Hajiseyedjavadi, O., Baase, W.A., and Matthews, B.W. 1993. The role of backbone flexibility in the accommodation of variants that repack the core of T4 lysozyme. Science 262 1715–1718. - PubMed
-
- Bornscheuer, U.T. and Pohl, M. 2001. Improved biocatalysts by directed evolution and rational protein design. Curr. Opin. Chem. Biol. 5 137–143. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
