

Connexin 43 (GJA1) mutations cause the pleiotropic phenotype of oculodentodigital dysplasia
- PMID: 12457340
- PMCID: PMC379233
- DOI: 10.1086/346090
Connexin 43 (GJA1) mutations cause the pleiotropic phenotype of oculodentodigital dysplasia
Abstract
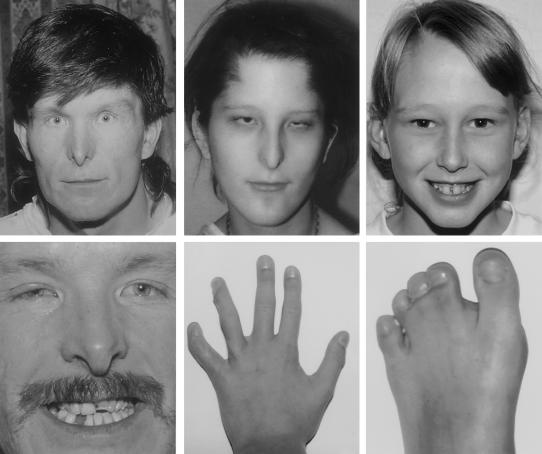
Gap junctions are assemblies of intercellular channels that regulate a variety of physiologic and developmental processes through the exchange of small ions and signaling molecules. These channels consist of connexin family proteins that allow for diversity of channel composition and conductance properties. The human connexin 43 gene, or GJA1, is located at human chromosome 6q22-q23 within the candidate region for the oculodentodigital dysplasia locus. This autosomal dominant syndrome presents with craniofacial (ocular, nasal, and dental) and limb dysmorphisms, spastic paraplegia, and neurodegeneration. Syndactyly type III and conductive deafness can occur in some cases, and cardiac abnormalities are observed in rare instances. We found mutations in the GJA1 gene in all 17 families with oculodentodigital dysplasia that we screened. Sixteen different missense mutations and one codon duplication were detected. These mutations may cause misassembly of channels or alter channel conduction properties. Expression patterns and phenotypic features of gja1 animal mutants, reported elsewhere, are compatible with the pleiotropic clinical presentation of oculodentodigital dysplasia.
Figures



References
Electronic-Database Information
-
- ClustalW, http://www.ebi.ac.uk/clustalw/ (for ClustalW program)
-
- CMT Mutations by Gene, http://molgen-www.uia.ac.be/CMTMutations/DataSource/MutByGene.cfm
-
- Connexin-Deafness Home Page, http://www.crg.es/deafness
-
- Entrez-Protein, http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?CMD=search&DB=protein (for accession numbers in )
-
- NCBI Sequence Viewer, http://www.ncbi.nlm.nih.gov/entrez/viewer.fcgi?val=22056350 (for human chromosome 6 sequence)
References
-
- Bergoffen J, Scherer SS, Wang S, Scott MO, Bone LJ, Paul DL, Chen K, Lensch MW, Chance PF, Fischbeck KH (1993) Connexin mutations in X-linked Charcot-Marie-Tooth disease. Science 262:2039–2042 - PubMed
-
- Boyadjiev SA, Jabs EW, LaBuda M, Jamal JE, Torbergsen T, Ptacek LJ 2nd, Rogers RC, Nyberg-Hansen R, Opjordsmoen S, Zeller CB, Stine OC, Stalker HJ, Zori RT, Shapiro RE (1999) Linkage analysis narrows the critical region for oculodentodigital dysplasia to chromosome 6q22-q23. Genomics 58:34–40 - PubMed
-
- Boyadjiev SA, Chowdry AB, Shapiro RE, Paznekas WA, Wandstrat AE, Choi JW, Kasch L, Zhang G, Wollnik B, Burgess CE, Schalling M, Lovett M, Jabs EW. Physical map of the chromosome 6q22 region containing the oculodentodigital dysplasia locus: analysis of thirteen candidate genes and identification of novel ESTs and DNA polymorphisms. Cytogenet Genome Res (in press) - PubMed
-
- Britz-Cunningham SH, Shah MM, Zuppan CW, Fletcher WH (1995) Mutations of the connexin43 gap-junction gene in patients with heart malformations and defects of laterality. N Engl J Med 332:1323–1329 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
