

NPHS2 mutations in late-onset focal segmental glomerulosclerosis: R229Q is a common disease-associated allele
- PMID: 12464671
- PMCID: PMC151634
- DOI: 10.1172/JCI16242
NPHS2 mutations in late-onset focal segmental glomerulosclerosis: R229Q is a common disease-associated allele
Abstract
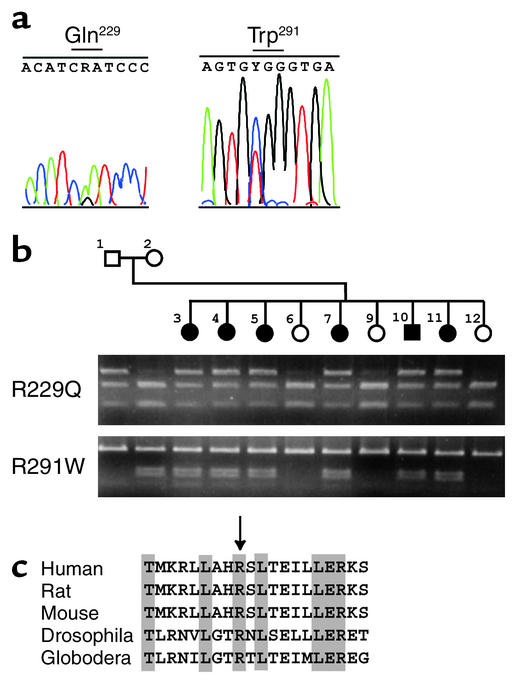
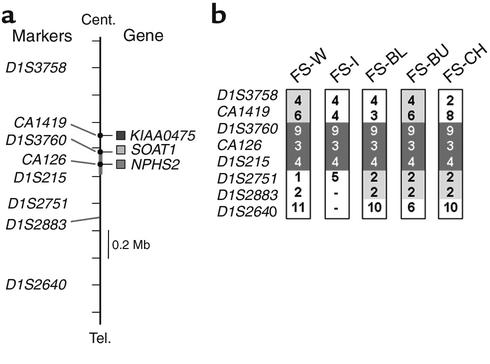
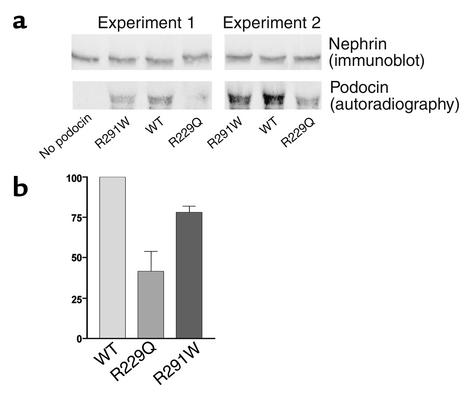
Mutations in NPHS2, encoding podocin, have been identified in childhood onset focal and segmental glomerulosclerosis (FSGS). The role of NPHS2 in adult disease is less well defined. We studied 30 families with FSGS and apparent autosomal recessive inheritance and 91 individuals with primary FSGS. We screened family members for NPHS2 mutations. NPHS2 mutations appeared to be responsible for disease in nine of these families. In six families, the affected individuals were compound heterozygotes for a nonconservative R229Q amino acid substitution. This R229Q variant has an allele frequency of 3.6% in a control population. In these families, R229Q was the only mutation identified on one of the two disease-associated NPHS2 alleles. We used in vitro-translated podocin and purified nephrin to investigate the effect of R229Q on their interaction and found decreased nephrin binding to the R229Q podocin. These data suggest that this common polymorphism contributes to the development of FSGS. Chromosomes bearing the R229Q mutation share a common haplotype defining an approximately 0.2-Mb region. R229Q appears to enhance susceptibility to FSGS in association with a second mutant NPHS2 allele. Identification of R229Q mutations may be of clinical importance, as NPHS2-associated disease appears to define a subgroup of FSGS patients unresponsive to corticosteroids.
Figures
References
-
- D’Agati V. The many masks of focal segmental glomerulosclerosis. Kidney Int. 1994;46:1223–1241. - PubMed
-
- Ichikawa I, Fogo A. Focal segmental glomerulsoclerosis. Pediatr Nephrol. 1996;10:374–391. - PubMed
-
- Somlo S, Mundel P. Getting a foothold in nephrotic syndrome. Nat Genet. 2000;24:333–335. - PubMed
-
- Kestila M, et al. Positionally cloned gene for a novel glomerular protein — nephrin — is mutated in congenital nephrotic syndrome. Mol Cell. 1998;1:575–582. - PubMed
-
- Holzman LB, et al. Nephrin localizes to the slit pore of the glomerular epithelial cell. Kidney Int. 1999;56:1481–1491. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
