

Interrogating a high-density SNP map for signatures of natural selection
- PMID: 12466284
- PMCID: PMC187574
- DOI: 10.1101/gr.631202
Interrogating a high-density SNP map for signatures of natural selection
Abstract
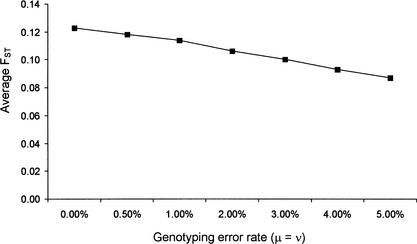
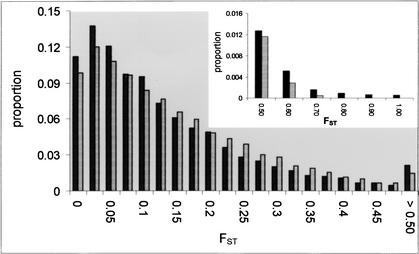
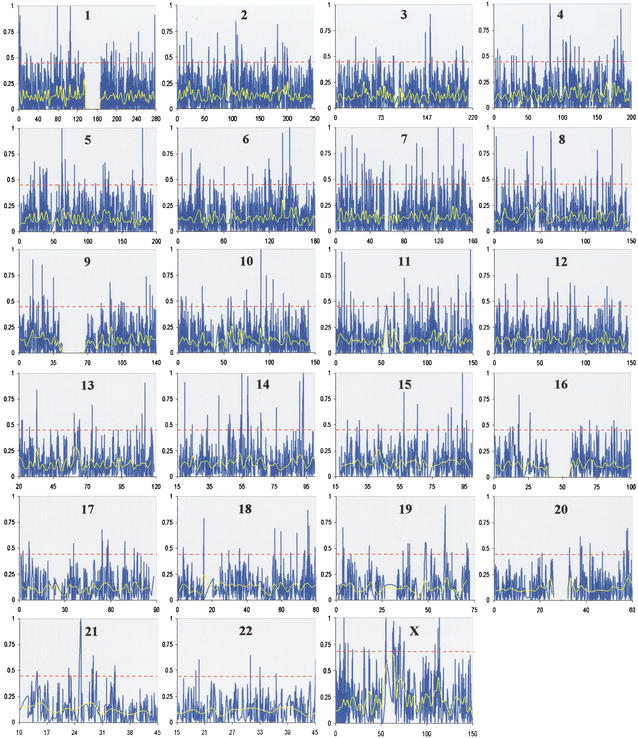
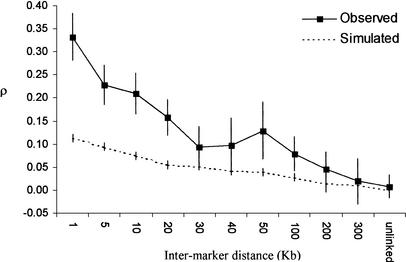
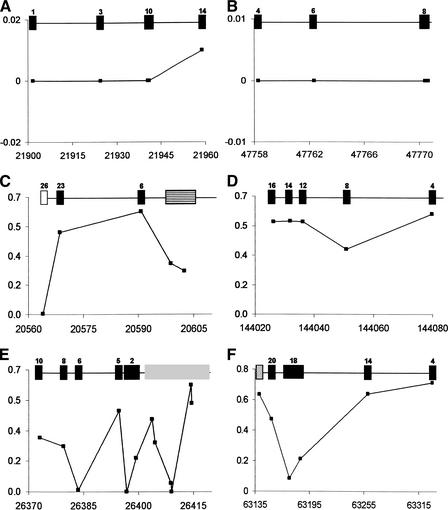
Identifying genomic regions that have been targets of natural selection remains one of the most important and challenging areas of research in genetics. To this end, we report an analysis of 26,530 single nucleotide polymorphisms (SNPs) with allele frequencies that were determined in three populations. Specifically, we calculated a measure of genetic differentiation, F(ST), for each locus and examined its distribution at the level of the genome, the chromosome, and individual genes. Through a variety of analyses, we have found statistically significant evidence supporting the hypothesis that selection has influenced extant patterns of human genetic variation. Importantly, by contrasting the F(ST) of individual SNPs to the empirical genome-wide distribution of F(ST), our results are not confounded by tenuous assumptions of population demographic history. Furthermore, we have identified 174 candidate genes with distribution of genetic variation that indicates that they have been targets of selection. Our work provides a first generation natural selection map of the human genome and provides compelling evidence that selection has shaped extant patterns of human genomic variation.
Figures
References
-
- Altshuler D, Pollar VJ, Cowles CR, Van Etten WJ, Baldwin J, Linton L, Lander ES. A SNP map of the human genome generated by reduced representation shotgun sequencing. Nature. 2000;407:513–516. - PubMed
-
- Andolfatto P. Adaptive hitchhiking effects on genome variability. Curr Opin Genet Dev. 2001;11:635–641. - PubMed
-
- Aquadro CF, Bauer DuMont V, Reed FA. Genome-wide variation in the human and fruitfly: A comparison. Curr Opin Genet Dev. 2001;1:627–634. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous