

A founding locus within the RET proto-oncogene may account for a large proportion of apparently sporadic Hirschsprung disease and a subset of cases of sporadic medullary thyroid carcinoma
- PMID: 12474140
- PMCID: PMC420016
- DOI: 10.1086/345466
A founding locus within the RET proto-oncogene may account for a large proportion of apparently sporadic Hirschsprung disease and a subset of cases of sporadic medullary thyroid carcinoma
Abstract
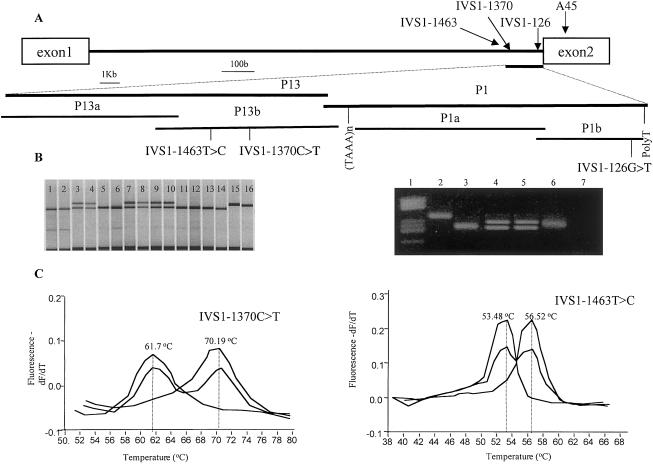
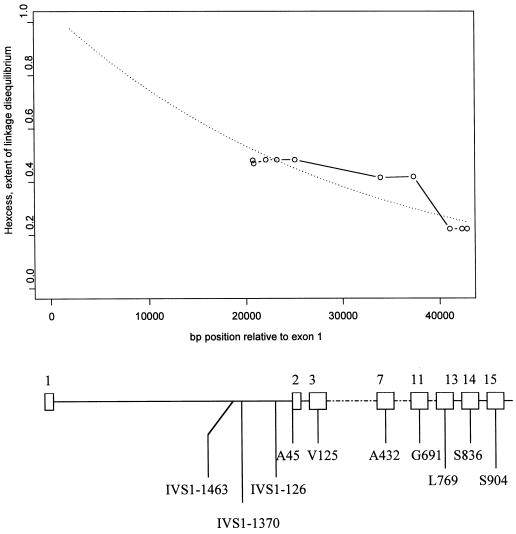
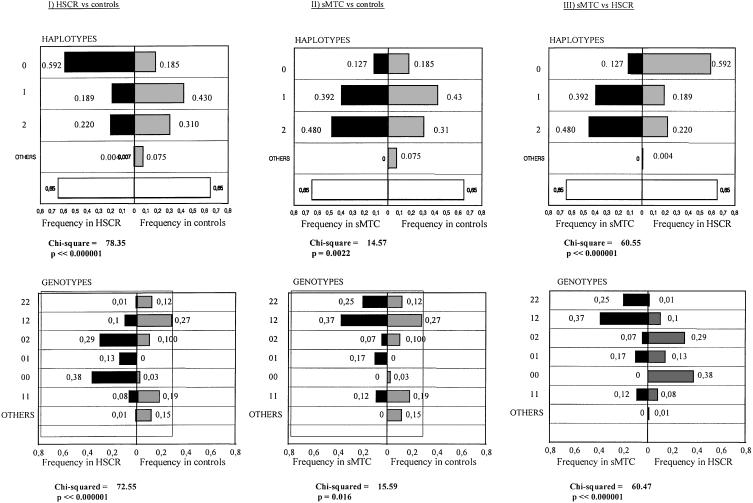
Hirschsprung disease (HSCR) is a common congenital disorder characterized by aganglionosis of the gut. The seemingly unrelated multiple endocrine neoplasia type 2 (MEN 2) is an autosomal dominant disorder characterized by medullary thyroid carcinoma (MTC), pheochromocytoma, and hyperparathyroidism. Yet, germline mutations in the RET proto-oncogene are associated with both MEN 2 and HSCR. In the former, gain-of-function mutations in a limited set of codons is found, whereas, in the latter, loss-of-function mutations are found. However, germline RET mutation is associated with only 3% of a population-based series of isolated HSCR, and little is known about susceptibility to sporadic MTC. We have found previously that specific haplotypes comprising RET coding single-nucleotide polymorphisms (SNPs) comprising exon 2 SNP A45A were strongly associated with HSCR, whereas haplotypes associated with exon 14 SNP S836S were associated with MTC. In this study, we describe three novel intron 1 SNPs, and, together with the coding SNP haplotypes, the data suggest the presence of distinct ancestral haplotypes for HSCR and sporadic MTC in linkage disequilibrium with a putative founding susceptibility locus/loci. The data are consistent with the presence of a very ancient, low-penetrance founder locus approximately 20-30 kb upstream of SNP A45A, but the failure of the SNPs to span the locus presents challenges in modeling mode of transmission or ancestry. We postulate that this founding locus is germane to both isolated HSCR and MTC but also that different mutations in this locus would predispose to one or the other.
Figures
References
Electronic-Database Information
-
- GenBank, http://www.ncbi.nlm.nih.gov/Genbank/index.html (for RET sequence [accession number AC010864])
-
- NCBI, http://www.ncbi.nlm.nih.gov/ (for Blast 2 sequences tool)
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for HSCR [MIM 142623] and MEN 2 [MIM 164761])
References
-
- Amiel J, Attié T, Jan D, Pelet A, Edery P, Bidaud C, Lacombe D, Tam P, Simeoni J, Flori E, Nihoul-Fékété C, Munnich A, Lyonnet S (1996) Heterozygous endothelin receptor B (EDNRB) mutations in isolated Hirschsprung disease. Hum Mol Genet 5:355–357 - PubMed
-
- Amiel J, Espinosa-Parilla Y, Steffan J, Gosset P, Pelet A, Prieru M, Boute O, Choiset A, Lacombe D, Philip N, Le Merrer M, Tanaka H, Till M, Touraine R, Toutain A, Vekemans M, Munnich A, Lyonnet S (2001) Large-scale deletions and SMADIP1 truncating mutations in syndromic Hirschsprung disease with involvement of midline structures. Am J Hum Genet 69:1370–1377 - PMC - PubMed
-
- Angrist M, Bolk S, Halushka M, Lapchak PA, Chakravarti A (1996) Germline mutations in glial cell line-derived neurotrophic factor (GDNF) and RET in a Hirschsprung disease patient. Nat Genet 14:341–344 - PubMed
-
- Angrist M, Bolk S, Thiel B, Puffenberger EG, Hofstra RM, Buys CHCM, Cass DT, Chakravarti A (1995) Mutation analysis of the RET receptor tyrosine kinase in Hirschsprung disease. Hum Mol Genet 4:821–830 - PubMed
-
- Attié T, Pelet A, Edery P, Eng C, Mulligan LM, Amiel J, Boutrand L, Beldjord C, Nihoul-Fékété C, Munnich A, Ponder BAJ, Lyonnet S (1995) Diversity of RET proto-oncogene mutations in familial and sporadic Hirschsprung disease. Hum Mol Genet 4:1381–1386 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
