

Aberrant neuronal and paracellular deposition of endostatin in brains of patients with Alzheimer's disease
- PMID: 12486154
- PMCID: PMC6758461
- DOI: 10.1523/JNEUROSCI.22-24-10621.2002
Aberrant neuronal and paracellular deposition of endostatin in brains of patients with Alzheimer's disease
Erratum in
- J Neurosci. 2003 Jan 15;23(2):725.
Abstract
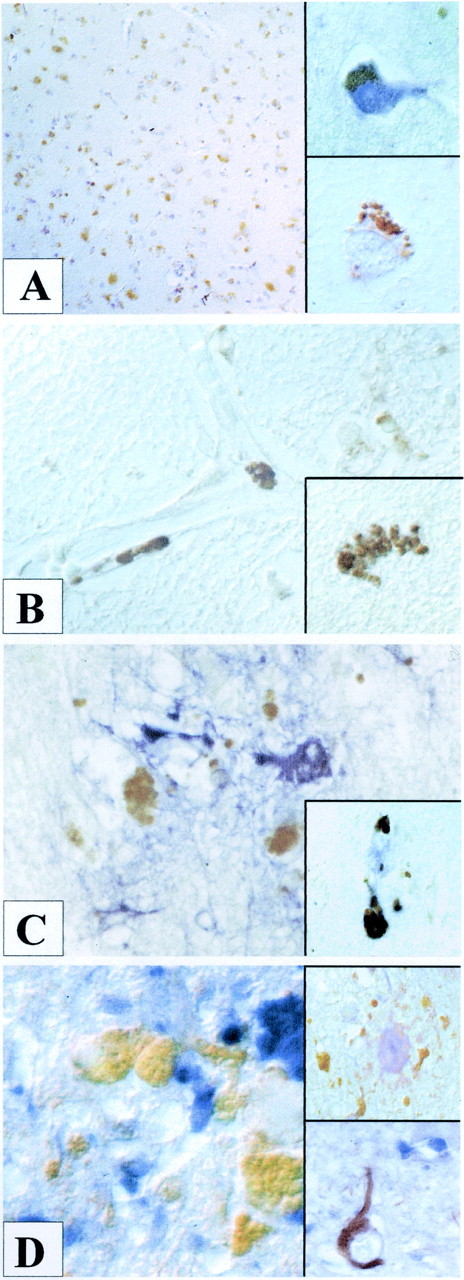
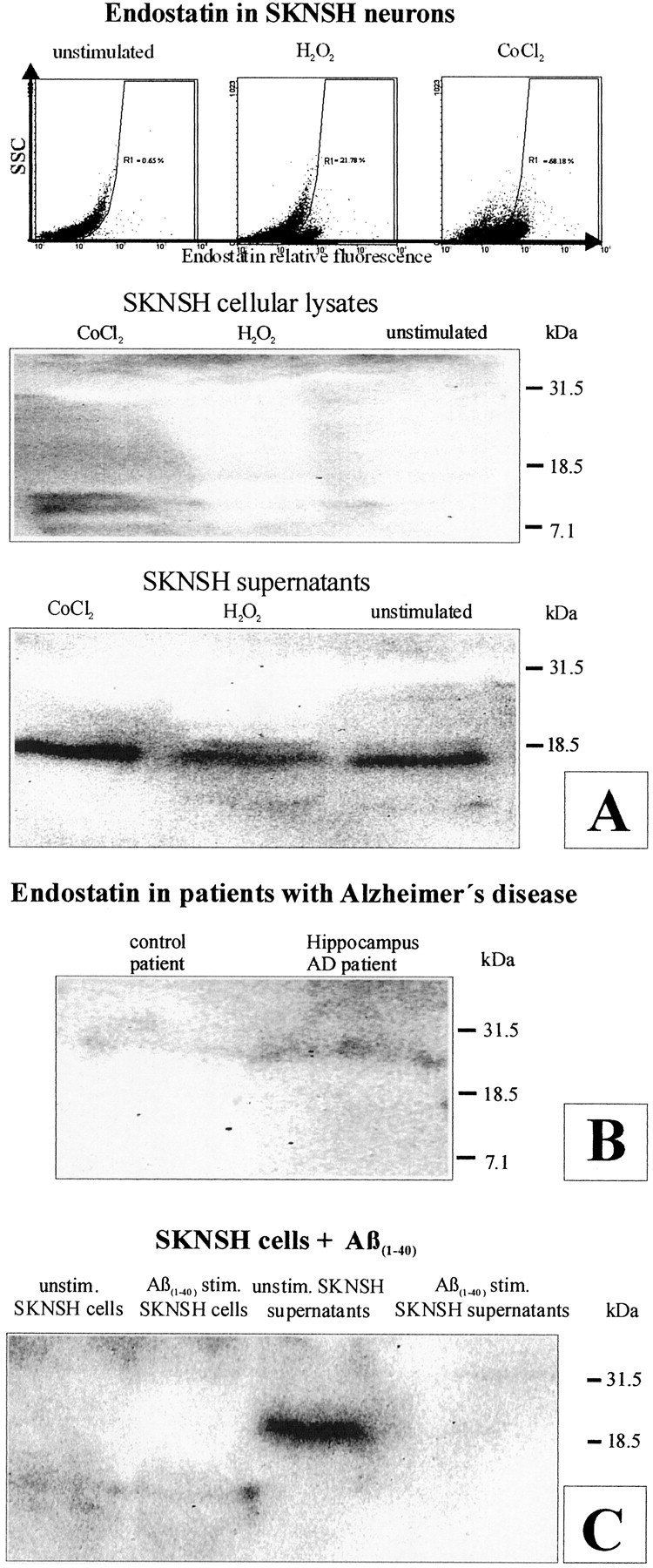
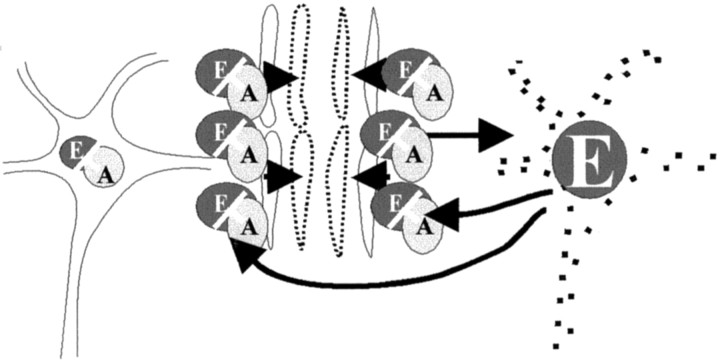
Cerebrovascular pathology is common in Alzheimer's disease (AD) and is considered to contribute to cerebral malfunction. However, distinct antiangiogenic proteins that accumulate in AD brains have not yet been identified. Endostatin is a 20 kDa C-terminal fragment of collagen XVIII that, when added exogenously, inhibits endothelial proliferation and migration in vitro and angiogenesis and tumor growth in vivo by inducing apoptosis in endothelial cells. We produced a monoclonal antibody directed against endostatin and observed significantly more (p < 0.0001) immunoreactive cortical neurons in AD brains compared with age-matched neuropathologically unaltered controls. High numbers of extracellular and frequently perivascular endostatin deposits were detected in the cerebral hemispheres. Double-labeling experiments revealed colocalization of endostatin in amyloid-beta(1-40) (Abeta(1-40)), tau protein, and periodic acid-Schiff stain-positive plaques that were surrounded by focal gliosis. Western blotting revealed more 20 kDa endostatin in an AD patient compared with a control. In unstimulated SKNSH supernatants, endostatin was detected that increased predominantly after hypoxia in supernatants and cellular lysates. Abeta(1-40) (80 microg/ml) supplementation to SKNSH neurons for 24 hr completely abolished the release of endostatin. These data show that endostatin is released by neurons to accumulate in amyloid plaques in Alzheimer's disease. Induction by hypoxia and complete abrogation of endostatin release after Abeta(1-40) challenge reveals intricate interactions between the two proteins and opens new avenues for the development of novel treatment strategies of AD patients.
Figures
Similar articles
-
Collagen XVIII: a novel heparan sulfate proteoglycan associated with vascular amyloid depositions and senile plaques in Alzheimer's disease brains.Brain Pathol. 2002 Oct;12(4):456-62. doi: 10.1111/j.1750-3639.2002.tb00462.x. Brain Pathol. 2002. PMID: 12408231 Free PMC article.
-
Endothelial endostatin release is induced by general cell stress and modulated by the nitric oxide/cGMP pathway.FASEB J. 2003 Jul;17(10):1267-76. doi: 10.1096/fj.02-1118com. FASEB J. 2003. PMID: 12832291
-
Accumulation of endostatin/collagenXVIII in brains of patients who died with cerebral malaria.J Neuroimmunol. 2002 Oct;131(1-2):216-21. doi: 10.1016/s0165-5728(02)00276-x. J Neuroimmunol. 2002. PMID: 12458056
-
Collagen XVIII/endostatin structure and functional role in angiogenesis.Cell Struct Funct. 2000 Apr;25(2):97-101. doi: 10.1247/csf.25.97. Cell Struct Funct. 2000. PMID: 10885579 Review.
-
Amyloid beta-peptide (1-42)-induced oxidative stress and neurotoxicity: implications for neurodegeneration in Alzheimer's disease brain. A review.Free Radic Res. 2002 Dec;36(12):1307-13. doi: 10.1080/1071576021000049890. Free Radic Res. 2002. PMID: 12607822 Review.
Cited by
-
Pharmacological approaches to improving cognitive function in Down syndrome: current status and considerations.Drug Des Devel Ther. 2014 Dec 17;9:103-25. doi: 10.2147/DDDT.S51476. eCollection 2015. Drug Des Devel Ther. 2014. PMID: 25552901 Free PMC article. Review.
-
VEGF receptors on PC12 cells mediate transient activation of ERK1/2 and Akt: comparison of nerve growth factor and vascular endothelial growth factor.J Negat Results Biomed. 2006 Jun 1;5:8. doi: 10.1186/1477-5751-5-8. J Negat Results Biomed. 2006. PMID: 16737552 Free PMC article.
-
Endostatin's emerging roles in angiogenesis, lymphangiogenesis, disease, and clinical applications.Biochim Biophys Acta. 2015 Dec;1850(12):2422-38. doi: 10.1016/j.bbagen.2015.09.007. Epub 2015 Sep 12. Biochim Biophys Acta. 2015. PMID: 26367079 Free PMC article. Review.
-
Impaired orthotopic glioma growth and vascularization in transgenic mouse models of Alzheimer's disease.J Neurosci. 2010 Aug 25;30(34):11251-8. doi: 10.1523/JNEUROSCI.2586-10.2010. J Neurosci. 2010. PMID: 20739545 Free PMC article.
-
The overlap between neurodegenerative and vascular factors in the pathogenesis of dementia.Acta Neuropathol. 2010 Sep;120(3):287-96. doi: 10.1007/s00401-010-0718-6. Epub 2010 Jul 11. Acta Neuropathol. 2010. PMID: 20623294 Free PMC article. Review.
References
-
- Bayer TA, Fossgreen A, Czech C, Beyreuther K, Wiestler OD. Plaque formation in brain transplants exposed to human beta-amyloid precursor protein 695. Acta Neuropathol Berl. 1996;92:130–137. - PubMed
-
- Beyreuther K, Bush AI, Dyrks T, Hilbich C, Konig G, Monning U, Multhaup G, Prior R, Rumble B, Schubert W. Mechanisms of amyloid deposition in Alzheimer's disease. Ann NY Acad Sci. 1991;640:129–139. - PubMed
-
- Bloch W, Huggel K, Sasaki T, Grose R, Bugnon P, Addicks K, Timpl R, Werner S. The angiogenesis inhibitor endostatin impairs blood vessel maturation during wound healing. FASEB J. 2000;14:2373–2376. - PubMed
-
- Boehm T, Folkman J, Browder T, O'Reilly MS. Antiangiogenic therapy of experimental cancer does not induce acquired drug resistance. Nature. 1997;390:404–407. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical