

Dihydropyridine receptors as voltage sensors for a depolarization-evoked, IP3R-mediated, slow calcium signal in skeletal muscle cells
- PMID: 12508050
- PMCID: PMC2217318
- DOI: 10.1085/jgp.20028671
Dihydropyridine receptors as voltage sensors for a depolarization-evoked, IP3R-mediated, slow calcium signal in skeletal muscle cells
Abstract
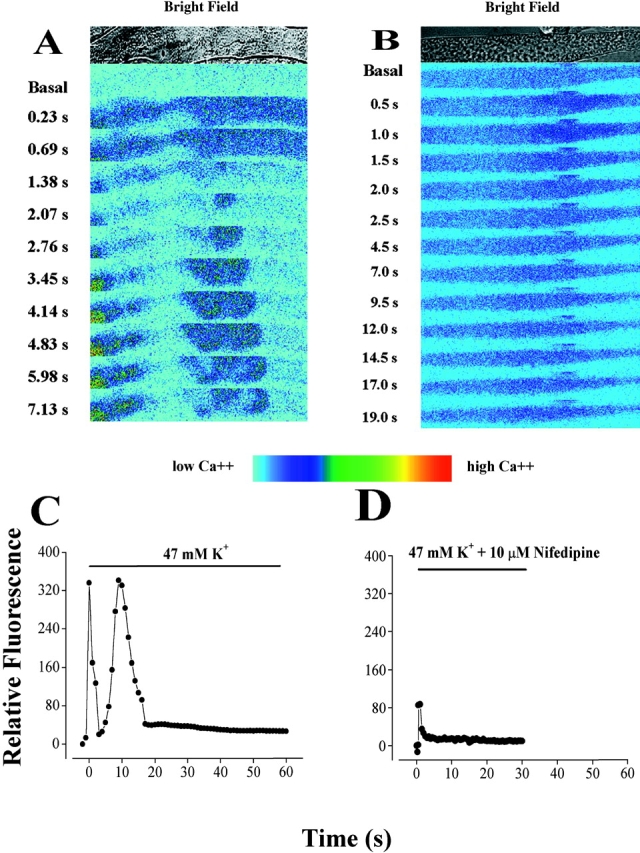
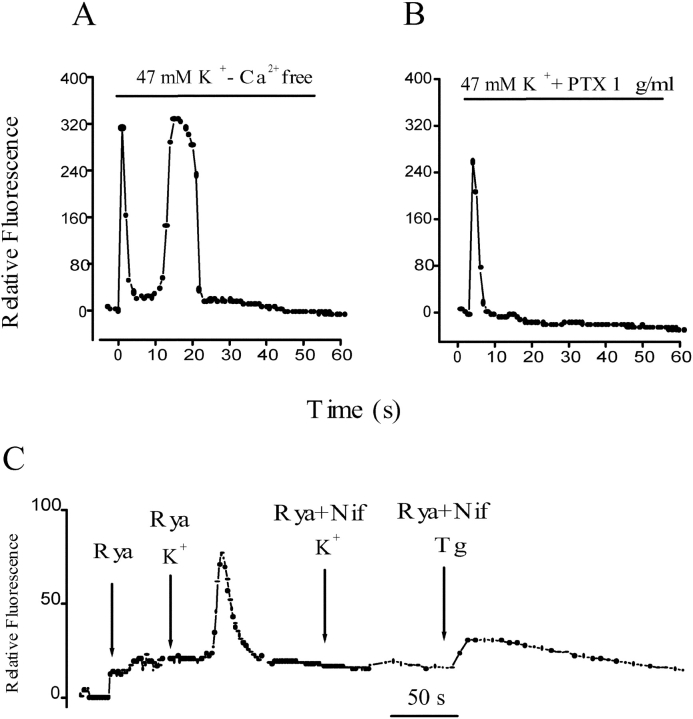
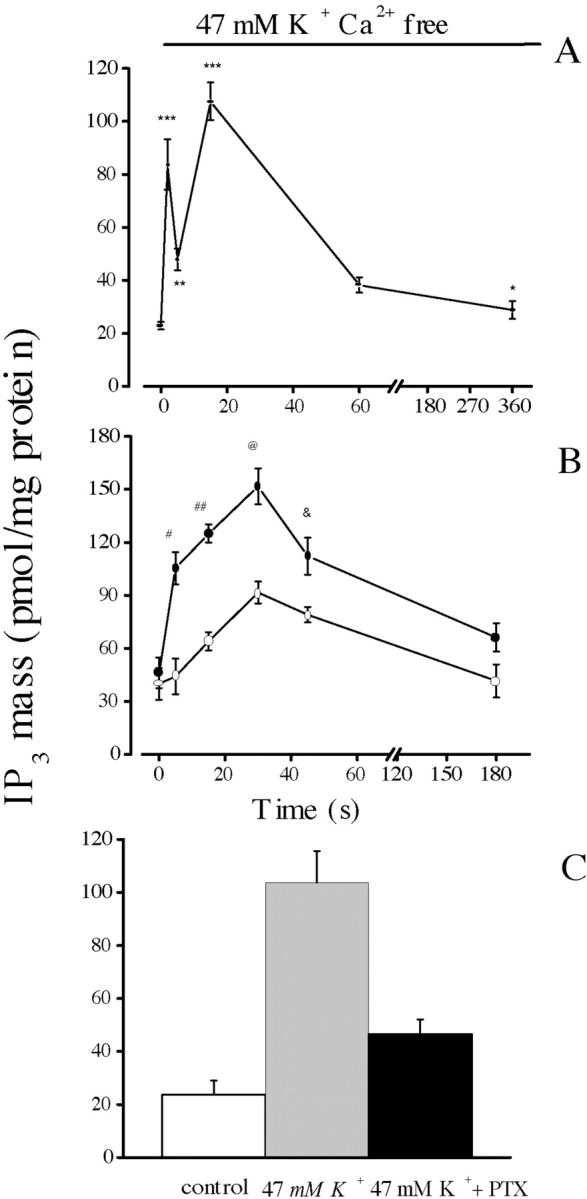
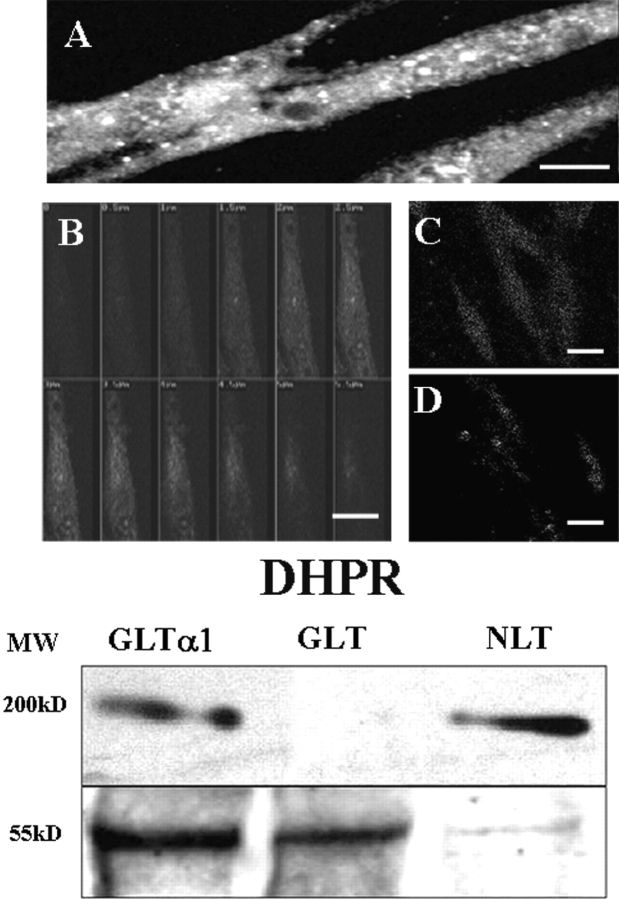
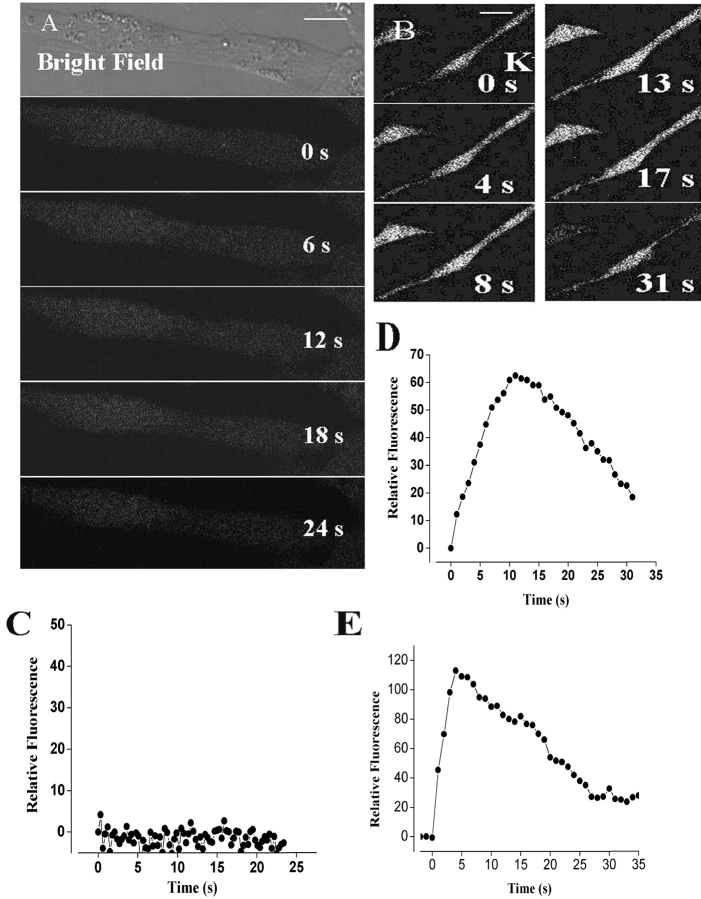
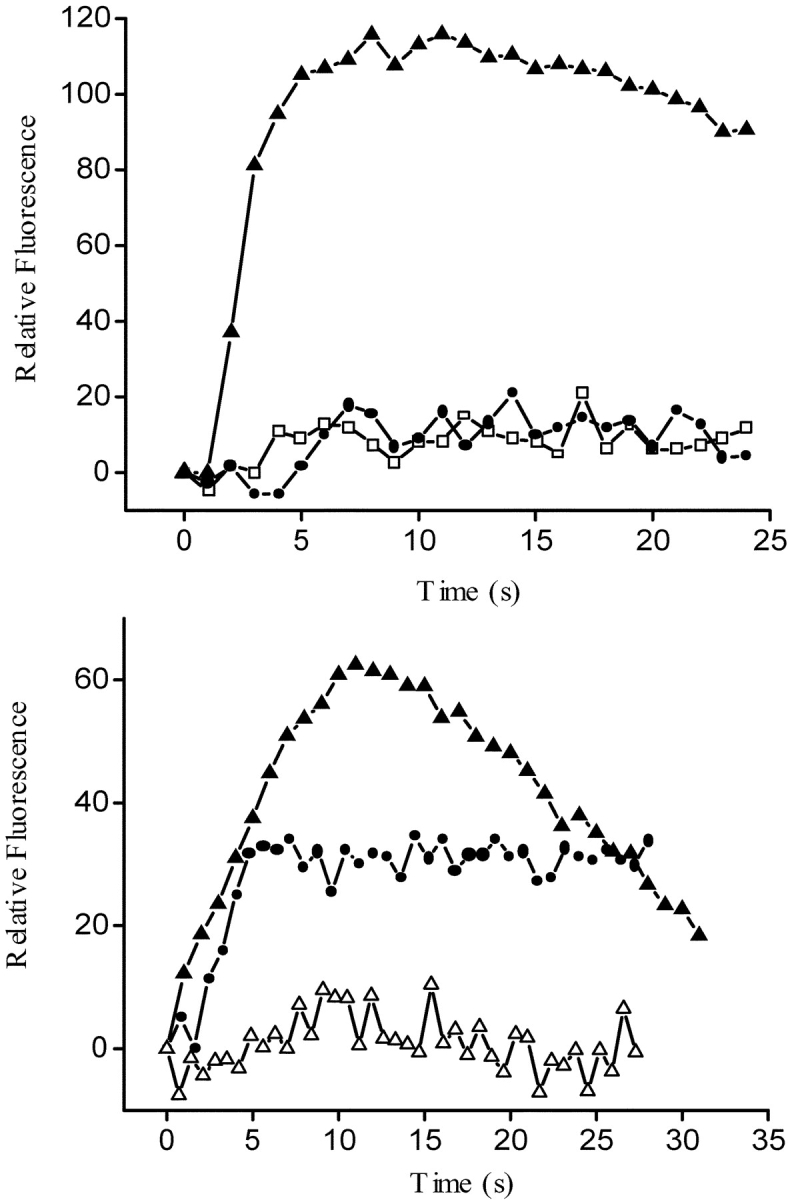
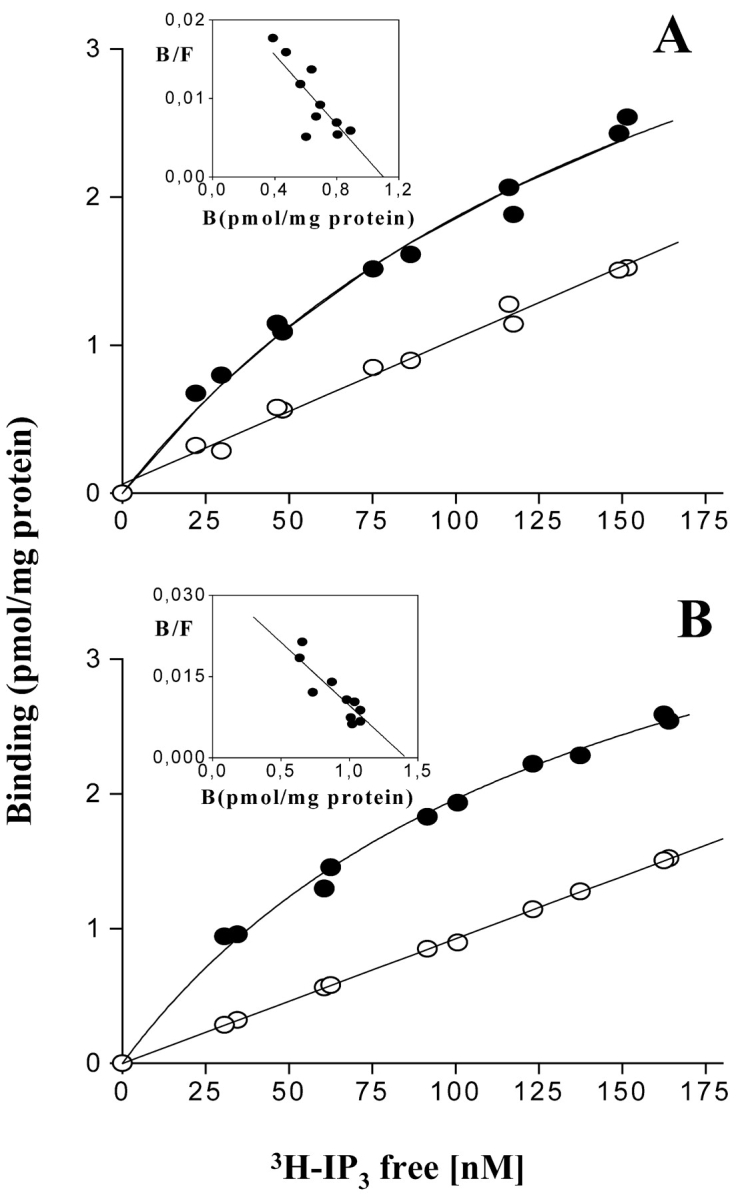
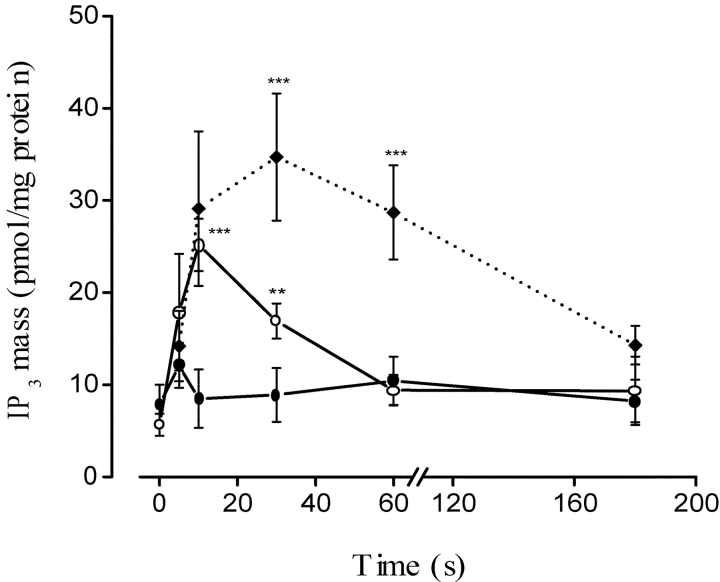
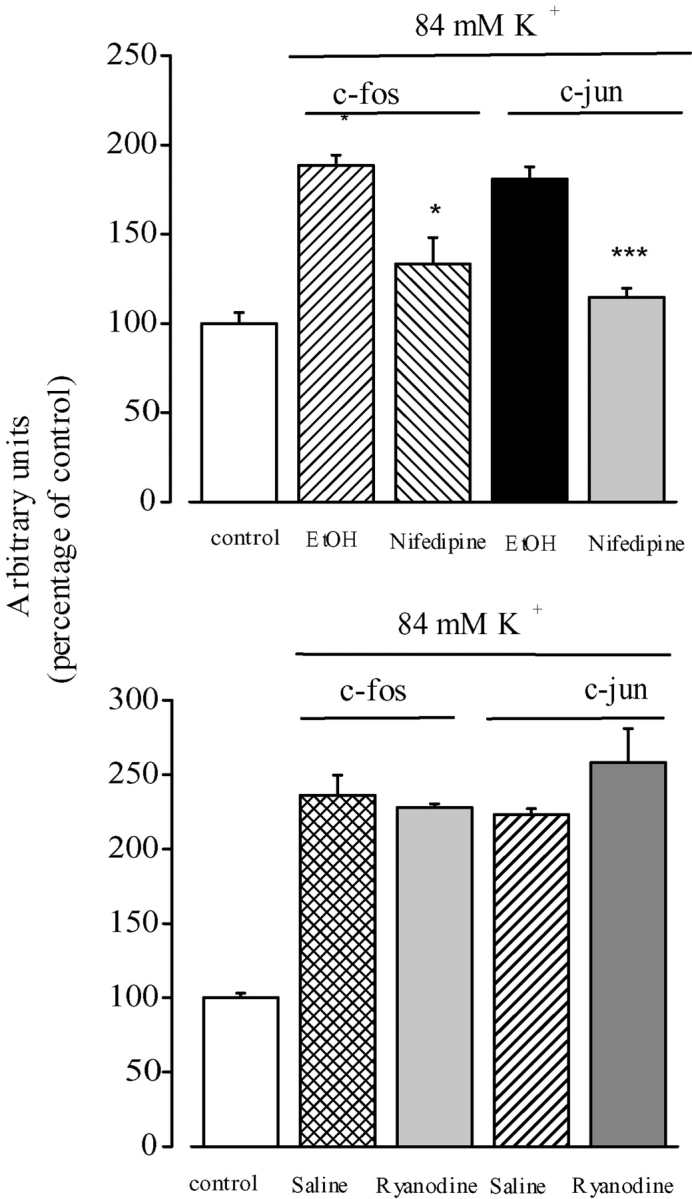
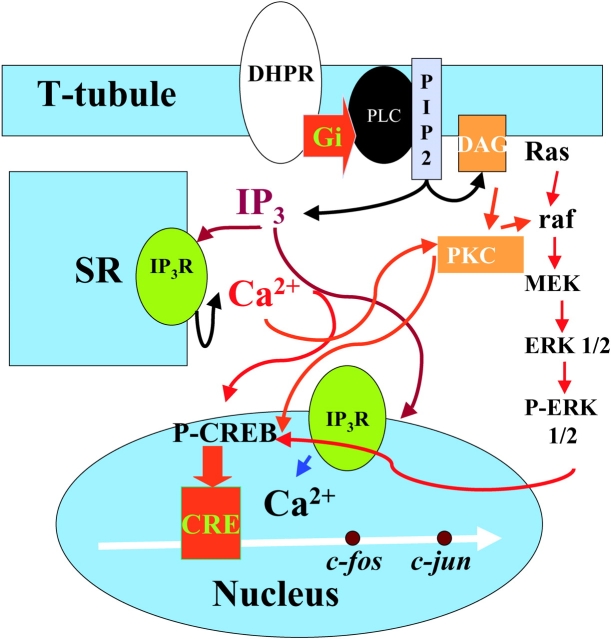
The dihydropyridine receptor (DHPR), normally a voltage-dependent calcium channel, functions in skeletal muscle essentially as a voltage sensor, triggering intracellular calcium release for excitation-contraction coupling. In addition to this fast calcium release, via ryanodine receptor (RYR) channels, depolarization of skeletal myotubes evokes slow calcium waves, unrelated to contraction, that involve the cell nucleus (Jaimovich, E., R. Reyes, J.L. Liberona, and J.A. Powell. 2000. Am. J. Physiol. Cell Physiol. 278:C998-C1010). We tested the hypothesis that DHPR may also be the voltage sensor for these slow calcium signals. In cultures of primary rat myotubes, 10 micro M nifedipine (a DHPR inhibitor) completely blocked the slow calcium (fluo-3-fluorescence) transient after 47 mM K(+) depolarization and only partially reduced the fast Ca(2+) signal. Dysgenic myotubes from the GLT cell line, which do not express the alpha(1) subunit of the DHPR, did not show either type of calcium transient following depolarization. After transfection of the alpha(1) DNA into the GLT cells, K(+) depolarization induced slow calcium transients that were similar to those present in normal C(2)C(12) and normal NLT cell lines. Slow calcium transients in transfected cells were blocked by nifedipine as well as by the G protein inhibitor, pertussis toxin, but not by ryanodine, the RYR inhibitor. Since slow Ca(2+) transients appear to be mediated by IP(3), we measured the increase of IP(3) mass after K(+) depolarization. The IP(3) transient seen in control cells was inhibited by nifedipine and was absent in nontransfected dysgenic cells, but alpha(1)-transfected cells recovered the depolarization-induced IP(3) transient. In normal myotubes, 10 micro M nifedipine, but not ryanodine, inhibited c-jun and c-fos mRNA increase after K(+) depolarization. These results suggest a role for DHPR-mediated calcium signals in regulation of early gene expression. A model of excitation-transcription coupling is presented in which both G proteins and IP(3) appear as important downstream mediators after sensing of depolarization by DHPR.
Figures
References
-
- Carrasco, M.A., N. Riveros, J. Ríos, and E. Jaimovich. 2000. Expression of early genes in K+-depolarized rat myotubes in primary culture. J. Physiol. 523P:100P.
-
- Chomczynski, P., and N. Sacchi. 1987. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 162:156–159. - PubMed
-
- Cognard, C., M. Rivet, and G. Raymond. 1990. The blockade of excitation/contraction coupling by nifedipine in patch-clamped rat skeletal muscle cells in culture. Pflugers Arch. 416:98–105. - PubMed
-
- Dolphin, A.C. 1999. L-Type calcium channel modulation. Adv. Second Messenger Phosphoprotein Res. 33:153–177. - PubMed
-
- Estrada, M., J.C. Cárdenas, J.L. Liberona, M.A. Carrasco, G.A. Mignery, P.D. Allen, and E. Jaimovich. 2001. Calcium transients in 1B5 myotubes lacking ryanodine receptors are related to inositol trisphosphate receptors. J. Biol. Chem. 276:22868–22874. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
