

Regulation of tumor necrosis factor alpha gene expression by mycobacteria involves the assembly of a unique enhanceosome dependent on the coactivator proteins CBP/p300
- PMID: 12509451
- PMCID: PMC151551
- DOI: 10.1128/MCB.23.2.526-533.2003
Regulation of tumor necrosis factor alpha gene expression by mycobacteria involves the assembly of a unique enhanceosome dependent on the coactivator proteins CBP/p300
Abstract
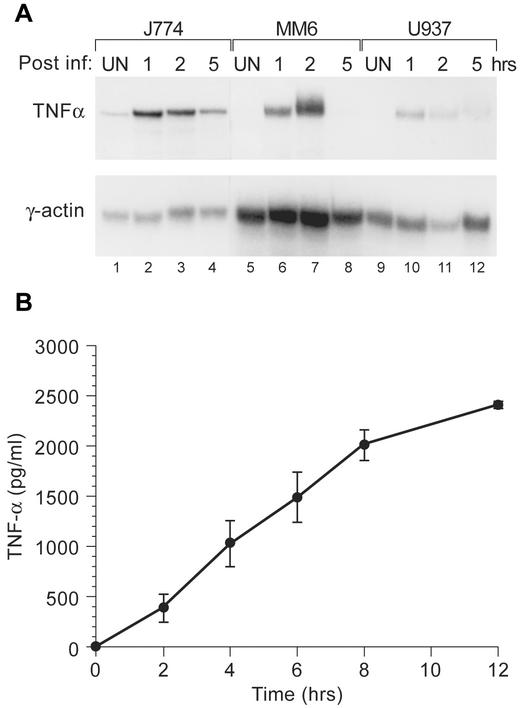
Tumor necrosis factor alpha (TNF-alpha) plays an important role in host containment of infection by Mycobacterium tuberculosis, one of the leading causes of death by an infectious agent globally. Using the pathogenic M. tuberculosis strain H37Rv, we present evidence that upon stimulation of monocytic cells by M. tuberculosis a unique TNF-alpha enhanceosome is formed, and it is distinct from the TNF-alpha enhanceosome that forms in T cells stimulated by antigen engagement or virus infection. A distinct set of activators including ATF-2, c-jun, Ets, Sp1, Egr-1 and the coactivator proteins CBP/p300 are recruited to the TNF-alpha promoter after stimulation with M. tuberculosis. Furthermore, the formation of this enhanceosome is dependent on inducer-specific helical phasing relationships between transcription factor binding sites. We also show that the transcriptional activity of CBP/p300 is potentiated by mycobacterial stimulation of monocytes. The identification of TNF-alpha regulatory elements and coactivators involved in M. tuberculosis-stimulated gene expression thus provides potential selective molecular targets in the modulation of TNF-alpha gene expression in the setting of mycobacterial infection.
Figures




References
-
- Bean, A. G., D. R. Roach, H. Briscoe, M. P. France, H. Korner, J. D. Sedgwick, and W. J. Britton. 1999. Structural deficiencies in granuloma formation in TNF gene-targeted mice underlie the heightened susceptibility to aerosol Mycobacterium tuberculosis infection, which is not compensated for by lymphotoxin. J. Immunol. 162:3504-3511. - PubMed
-
- Bermudez, L. E., and L. S. Young. 1988. Tumor necrosis factor, alone or in combination with IL-2, but not IFN-gamma, is associated with macrophage killing of Mycobacterium avium complex. J. Immunol. 140:3006-3013. - PubMed
-
- Carey, M. 1998. The enhanceosome and transcriptional synergy. Cell 92:5-8. - PubMed
-
- Chrivia, J. C., R. P. Kwok, N. Lamb, M. Hagiwara, M. R. Montminy, and R. H. Goodman. 1993. Phosphorylated CREB binds specifically to the nuclear protein CBP. Nature 365:855-859. - PubMed
-
- Eckner, R., M. E. Ewen, D. Newsome, M. Gerdes, J. A. DeCaprio, J. B. Lawrence, and D. M. Livingston. 1994. Molecular cloning and functional analysis of the adenovirus E1A-associated 300-kD protein (p300) reveals a protein with properties of a transcriptional adaptor. Genes Dev. 8:869-884. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous