

Role of ALDP (ABCD1) and mitochondria in X-linked adrenoleukodystrophy
- PMID: 12509471
- PMCID: PMC151532
- DOI: 10.1128/MCB.23.2.744-753.2003
Role of ALDP (ABCD1) and mitochondria in X-linked adrenoleukodystrophy
Abstract
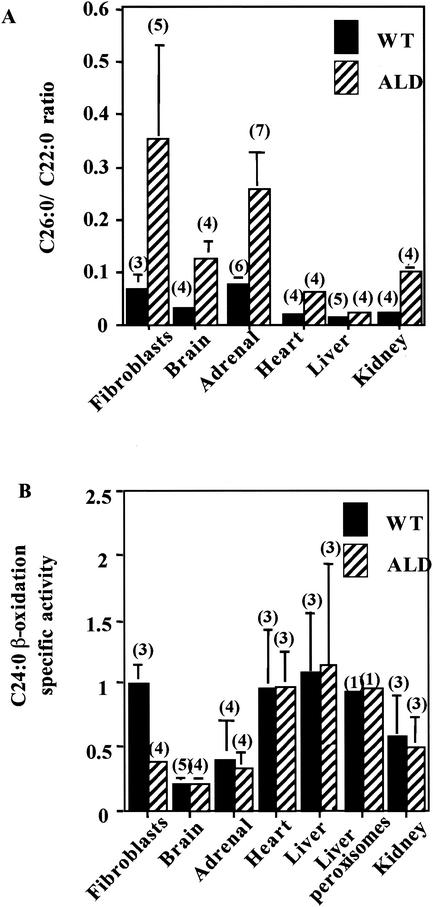
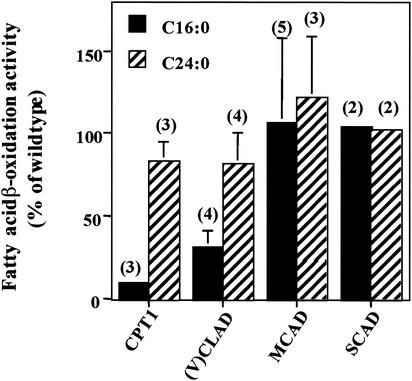
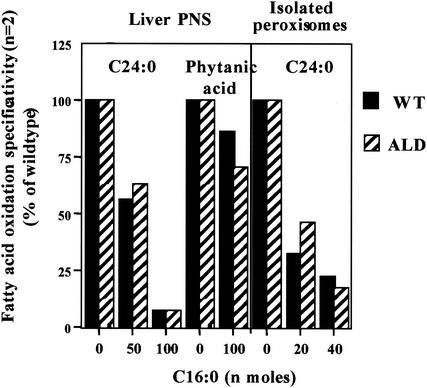
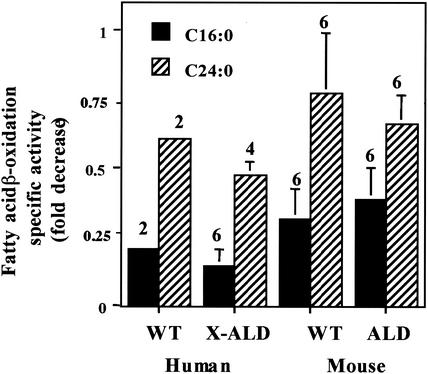
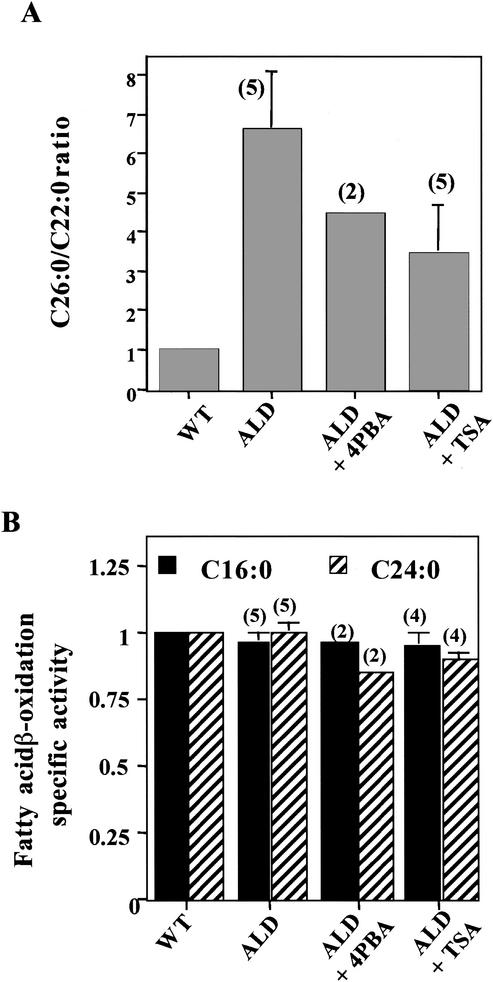
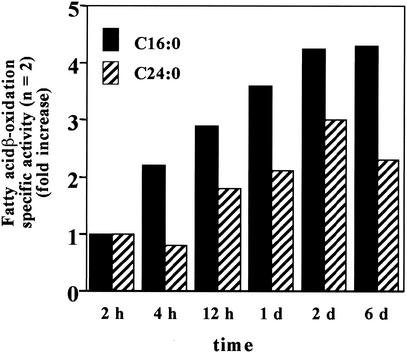
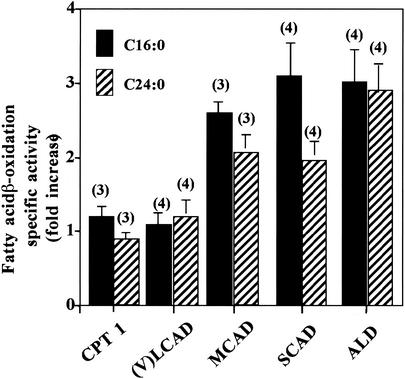
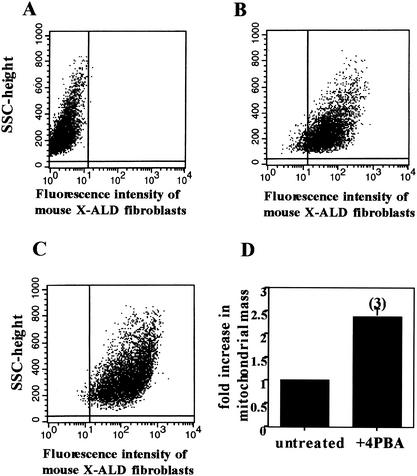
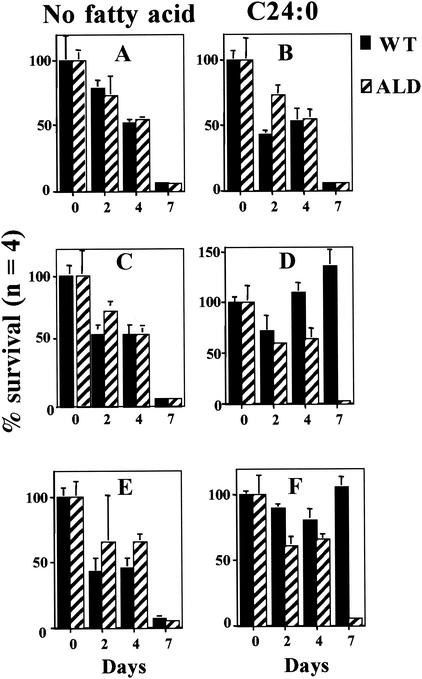
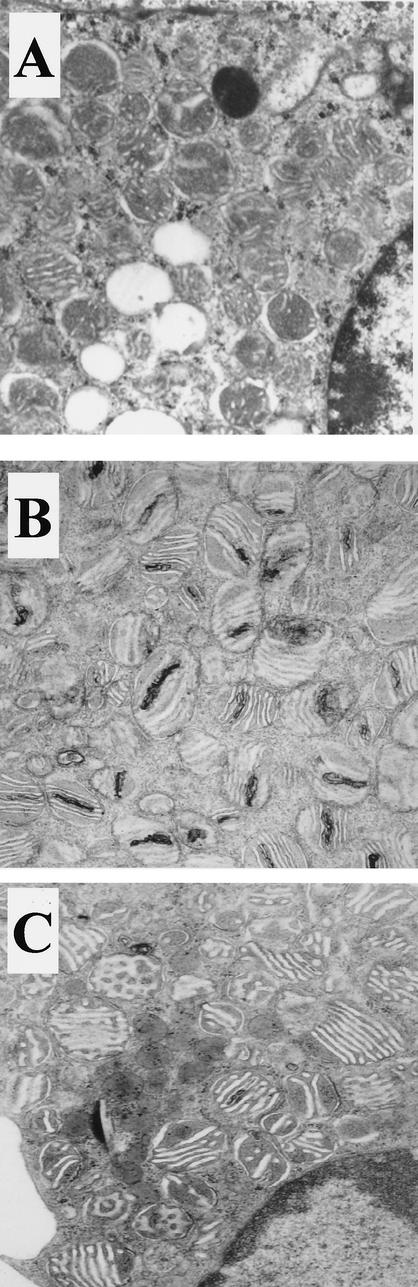
Peroxisomal disorders have been associated with malfunction of peroxisomal metabolic pathways, but the pathogenesis of these disorders is largely unknown. X-linked adrenoleukodystrophy (X-ALD) is associated with elevated levels of very-long-chain fatty acids (VLCFA; C(>22:0)) that have been attributed to reduced peroxisomal VLCFA beta-oxidation activity. Previously, our laboratory and others have reported elevated VLCFA levels and reduced peroxisomal VLCFA beta-oxidation in human and mouse X-ALD fibroblasts. In this study, we found normal levels of peroxisomal VLCFA beta-oxidation in tissues from ALD mice with elevated VLCFA levels. Treatment of ALD mice with pharmacological agents resulted in decreased VLCFA levels without a change in VLCFA beta-oxidation activity. These data indicate that ALDP does not determine the rate of VLCFA beta-oxidation and that VLCFA levels are not determined by the rate of VLCFA beta-oxidation. The rate of peroxisomal VLCFA beta-oxidation in human and mouse fibroblasts in vitro is affected by the rate of mitochondrial long-chain fatty acid beta-oxidation. We hypothesize that ALDP facilitates the interaction between peroxisomes and mitochondria, resulting, when ALDP is deficient in X-ALD, in increased VLCFA accumulation despite normal peroxisomal VLCFA beta-oxidation in ALD mouse tissues. In support of this hypothesis, mitochondrial structural abnormalities were observed in adrenal cortical cells of ALD mice.
Figures
References
-
- Allen, J., T. Kepic, D. Garwicki, and M. Yunus. 1982. Adrenal defect in adrenomyelodystrophy. South. Med. J. 75:877-879. - PubMed
-
- Berger, J., S. Albet, M. Bentejac, N. Netik, A. Holzinger, A. A. Roscher, M. Bugaut, and S. Forss-Petter. 1999. The four murine peroxisomal ABC-transporter genes differ in constitutive, inducible and developmental expression. Eur. J. Biochem. 265:719-727. - PubMed
-
- Bezman, L., A. Moser, G. Raymond, P. Rinaldo, P. Watkins, K. Smith, N. Kass, and H. Moser. 2001. Adrenoleukodystrophy: incidence, new mutation rate, and results of extended family screening. Ann. Neurol. 49:512-517. - PubMed
-
- Bissler, J. J., M. Tsoras, H. H. H. Goring, P. Hug, G. Chuck, E. Tombragel, C. McGraw, J. Schlotman, M. A. Ralston, and G. Hug. 2002. Infantile dilated X-linked cardiomyopathy, G4.5 mutations, altered lipids, and ultrastructural malformations of mitochondria in heart, liver, and skeletal muscle. Lab. Investig. 82:335-344. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases