

AVID: A global alignment program
- PMID: 12529311
- PMCID: PMC430967
- DOI: 10.1101/gr.789803
AVID: A global alignment program
Abstract
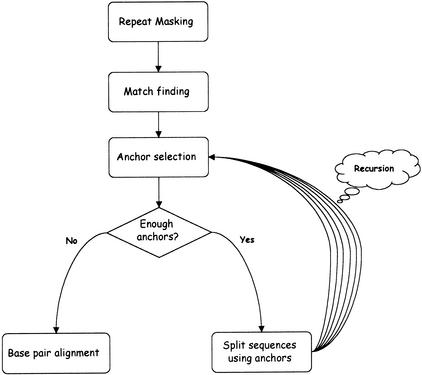
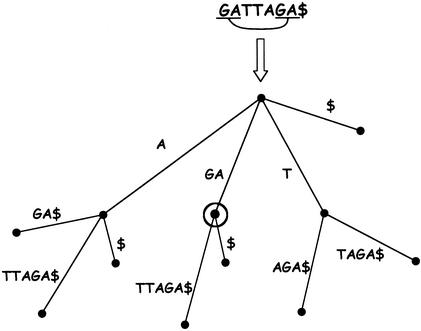
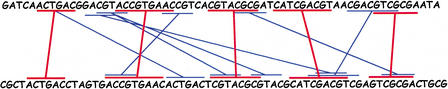
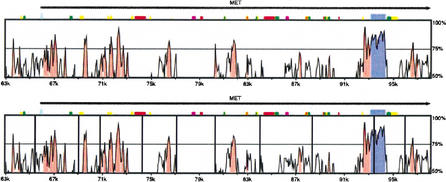
In this paper we describe a new global alignment method called AVID. The method is designed to be fast, memory efficient, and practical for sequence alignments of large genomic regions up to megabases long. We present numerous applications of the method, ranging from the comparison of assemblies to alignment of large syntenic genomic regions and whole genome human/mouse alignments. We have also performed a quantitative comparison of AVID with other popular alignment tools. To this end, we have established a format for the representation of alignments and methods for their comparison. These formats and methods should be useful for future studies. The tools we have developed for the alignment comparisons, as well as the AVID program, are publicly available. See Web Site References section for AVID Web address and Web addresses for other programs discussed in this paper.
Figures
References
-
- Altschul S.F., Gish, W., Miller, W., Myers, E.W., and Lipman, D.J. 1990. Basic local alignment search tool. J. Mol. Biol. 215: 403-410. - PubMed
-
- Benos P.V., Gatt, M.K., Murphy, L., Harris, D., Barrell, B., Ferraz, C., Vidal, S., Brun, C., Demaille, J., Cadieu, E., et al. 2001. From first base: The sequence of the tip of the X chromosome of Drosophila melanogaster, a comparison of two sequencing strategies. Genome Res. 11: 710-730. - PMC - PubMed
-
- Brudno M. and Morgenstern, B., 2002. Fast and sensitive alignment of large genomic sequences. In Proceedings of the First IEEE Computer Society Conference on Bioinformatics. IEEE Computer Society Press. - PubMed
-
- Burset M. and Guigo, R. 1996. Evaluation of gene structure prediction programs. Genomics 34: 353-357. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources