

Idiopathic restrictive cardiomyopathy is part of the clinical expression of cardiac troponin I mutations
- PMID: 12531876
- PMCID: PMC151864
- DOI: 10.1172/JCI16336
Idiopathic restrictive cardiomyopathy is part of the clinical expression of cardiac troponin I mutations
Erratum in
- J Clin Invest. 2003 Mar;111(6):925
Abstract
Restrictive cardiomyopathy (RCM) is an uncommon heart muscle disorder characterized by impaired filling of the ventricles with reduced volume in the presence of normal or near normal wall thickness and systolic function. The disease may be associated with systemic disease but is most often idiopathic. We recognized a large family in which individuals were affected by either idiopathic RCM or hypertrophic cardiomyopathy (HCM). Linkage analysis to selected sarcomeric contractile protein genes identified cardiac troponin I (TNNI3) as the likely disease gene. Subsequent mutation analysis revealed a novel missense mutation, which cosegregated with the disease in the family (lod score: 4.8). To determine if idiopathic RCM is part of the clinical expression of TNNI3 mutations, genetic investigations of the gene were performed in an additional nine unrelated RCM patients with restrictive filling patterns, bi-atrial dilatation, normal systolic function, and normal wall thickness. TNNI3 mutations were identified in six of these nine RCM patients. Two of the mutations identified in young individuals were de novo mutations. All mutations appeared in conserved and functionally important domains of the gene. This article was published online in advance of the print edition. The date of publication is available from the JCI website, http://www.jci.org.
Figures

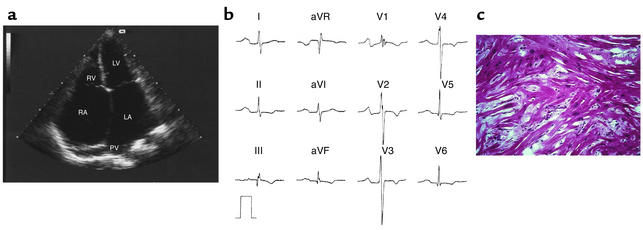

Comment on
-
Genotype, phenotype: upstairs, downstairs in the family of cardiomyopathies.J Clin Invest. 2003 Jan;111(2):175-8. doi: 10.1172/JCI17612. J Clin Invest. 2003. PMID: 12531871 Free PMC article. No abstract available.
References
-
- Richardson P, et al. Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the Definition and Classification of cardiomyopathies. Circulation. 1996;93:841–842. - PubMed
-
- Rivenes SM, Kearney DL, Smith EO, Towbin JA, Denfield SW. Sudden death and cardiovascular collapse in children with restrictive cardiomyopathy. Circulation. 2002;102:876–882. - PubMed
-
- Siegel RJ, Shah PK, Fishbein MC. Idiopathic restrictive cardiomyopathy. Circulation. 1984;70:165–169. - PubMed
-
- Keren A, Popp RL. Assignment of patients into the classification of cardiomyopathies. Circulation. 1992;86:1622–1633. - PubMed
-
- Kushwaha SS, Fallon JT, Fuster V. Restrictive cardiomyopathy. N. Engl. J. Med. 1997;336:267–276. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
