

Aberrant activation of focal adhesion proteins mediates fibrillar amyloid beta-induced neuronal dystrophy
- PMID: 12533609
- PMCID: PMC6741895
- DOI: 10.1523/JNEUROSCI.23-02-00493.2003
Aberrant activation of focal adhesion proteins mediates fibrillar amyloid beta-induced neuronal dystrophy
Abstract
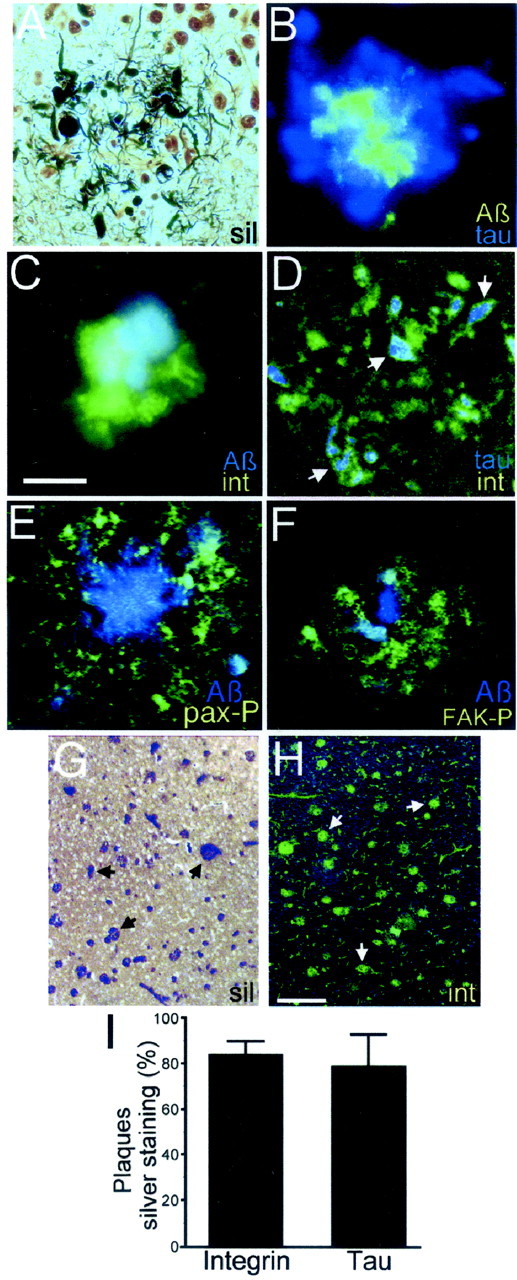
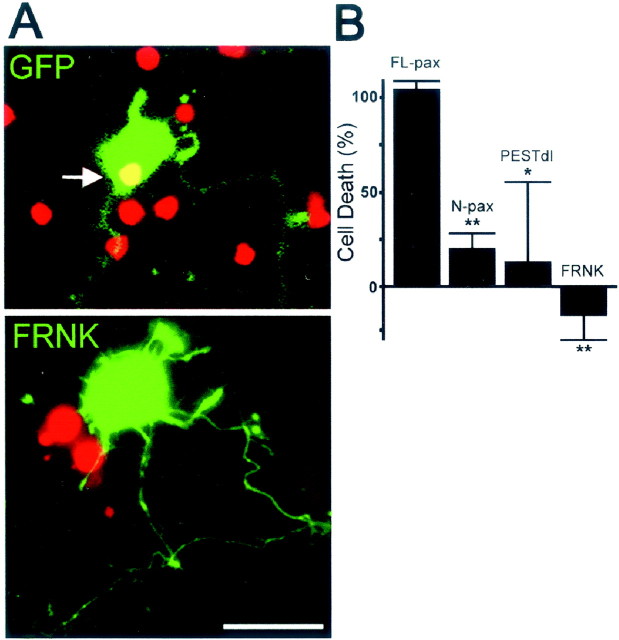
Neuronal dystrophy is a pathological hallmark of Alzheimer's disease (AD) that is not observed in other neurodegenerative disorders that lack amyloid deposition. Treatment of cortical neurons with fibrillar amyloid beta (Abeta) peptides induces progressive neuritic dystrophy accompanied by a marked loss of synaptophysin immunoreactivity (Grace et al., 2002). Here, we report that fibrillar Abeta-induced neuronal dystrophy is mediated by the activation of focal adhesion (FA) proteins and the formation of aberrant FA structures adjacent to Abeta deposits. In the AD brain, activated FA proteins are observed associated with the majority of senile plaques. Clustered integrin receptors and activated paxillin (phosphorylated at Tyr-31) and focal adhesion kinase (phosphorylated at Tyr-297) are mainly detected in dystrophic neurites surrounding Abeta plaque cores, where they colocalize with hyperphosphorylated tau. Deletion experiments demonstrated that the presence of the LIM domains in the paxillin C terminus and the recruitment of the protein-Tyr phosphatase (PTP)-PEST to the FA complex are required for Abeta-induced neuronal dystrophy. Therefore, both paxillin and PTP-PEST appear to be critical elements in the generation of the dystrophic response. Paxillin is a scaffolding protein to which other FA proteins bind, leading to the formation of the FA contact and initiation of signaling cascades. PTP-PEST plays a key role in the dynamic regulation of focal adhesion contacts in response to extracellular cues. Thus, in the AD brain, fibrillar Abeta may induce neuronal dystrophy by triggering a maladaptive plastic response mediated by FA protein activation and tau hyperphosphorylation.
Figures





Similar articles
-
Characterization of neuronal dystrophy induced by fibrillar amyloid beta: implications for Alzheimer's disease.Neuroscience. 2002;114(1):265-73. doi: 10.1016/s0306-4522(02)00241-5. Neuroscience. 2002. PMID: 12207971
-
Phosphorylation of actin-depolymerizing factor/cofilin by LIM-kinase mediates amyloid beta-induced degeneration: a potential mechanism of neuronal dystrophy in Alzheimer's disease.J Neurosci. 2006 Jun 14;26(24):6533-42. doi: 10.1523/JNEUROSCI.5567-05.2006. J Neurosci. 2006. PMID: 16775141 Free PMC article.
-
Rapid tyrosine phosphorylation of neuronal proteins including tau and focal adhesion kinase in response to amyloid-beta peptide exposure: involvement of Src family protein kinases.J Neurosci. 2002 Jan 1;22(1):10-20. doi: 10.1523/JNEUROSCI.22-01-00010.2002. J Neurosci. 2002. PMID: 11756483 Free PMC article.
-
Focal adhesions regulate Abeta signaling and cell death in Alzheimer's disease.Biochim Biophys Acta. 2007 Apr;1772(4):438-45. doi: 10.1016/j.bbadis.2006.11.007. Epub 2006 Nov 30. Biochim Biophys Acta. 2007. PMID: 17215111 Free PMC article. Review.
-
Aβ Influences Cytoskeletal Signaling Cascades with Consequences to Alzheimer's Disease.Mol Neurobiol. 2015 Dec;52(3):1391-1407. doi: 10.1007/s12035-014-8913-4. Epub 2014 Oct 26. Mol Neurobiol. 2015. PMID: 25344315 Review.
Cited by
-
The significance of neuroinflammation in understanding Alzheimer's disease.J Neural Transm (Vienna). 2006 Nov;113(11):1685-95. doi: 10.1007/s00702-006-0575-6. Epub 2006 Oct 13. J Neural Transm (Vienna). 2006. PMID: 17036175 Review.
-
Amyloid precursor protein (APP) and amyloid β (Aβ) interact with cell adhesion molecules: Implications in Alzheimer's disease and normal physiology.Front Cell Dev Biol. 2022 Jul 26;10:969547. doi: 10.3389/fcell.2022.969547. eCollection 2022. Front Cell Dev Biol. 2022. PMID: 35959488 Free PMC article. Review.
-
Kihi-to, a herbal traditional medicine, improves Abeta(25-35)-induced memory impairment and losses of neurites and synapses.BMC Complement Altern Med. 2008 Aug 16;8:49. doi: 10.1186/1472-6882-8-49. BMC Complement Altern Med. 2008. PMID: 18706097 Free PMC article.
-
Alzheimer's, Parkinson's Disease and Amyotrophic Lateral Sclerosis Gene Expression Patterns Divergence Reveals Different Grade of RNA Metabolism Involvement.Int J Mol Sci. 2020 Dec 14;21(24):9500. doi: 10.3390/ijms21249500. Int J Mol Sci. 2020. PMID: 33327559 Free PMC article.
-
Cytoskeletal pathologies of Alzheimer disease.Cell Motil Cytoskeleton. 2009 Aug;66(8):635-49. doi: 10.1002/cm.20388. Cell Motil Cytoskeleton. 2009. PMID: 19479823 Free PMC article. Review.
References
-
- Akiyama H, Kawamata T, Dedhar S, McGeer PL. Immunohistochemical localization of vitronectin, its receptor and beta-3 integrin in Alzheimer brain tissue. J Neuroimmunol. 1991;32:19–28. - PubMed
-
- Aplin AE, Howe A, Alahari SK, Juliano RL. Signal transduction and signal modulation by cell adhesion receptors: the role of integrins, cadherins, immunoglobulin-cell adhesion molecules, and selectins. Pharmacol Rev. 1998;50:197–263. - PubMed
-
- Baumann K, Mandelkow EM, Biernat J, Piwnica-Worms H, Mandelkow E. Abnormal Alzheimer-like phosphorylation of tau-protein by cyclin-dependent kinases cdk2 and cdk5. FEBS Lett. 1993;336:417–424. - PubMed
-
- Benzing WC, Mufson EJ, Armstrong DM. Alzheimer's disease-like dystrophic neurites characteristically associated with senile plaques are not found within other neurodegenerative diseases unless amyloid beta-protein deposition is present. Brain Res. 1993;606:10–18. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases