

Assessing the impact of comparative genomic sequence data on the functional annotation of the Drosophila genome
- PMID: 12537575
- PMCID: PMC151188
- DOI: 10.1186/gb-2002-3-12-research0086
Assessing the impact of comparative genomic sequence data on the functional annotation of the Drosophila genome
Abstract
Background: It is widely accepted that comparative sequence data can aid the functional annotation of genome sequences; however, the most informative species and features of genome evolution for comparison remain to be determined.
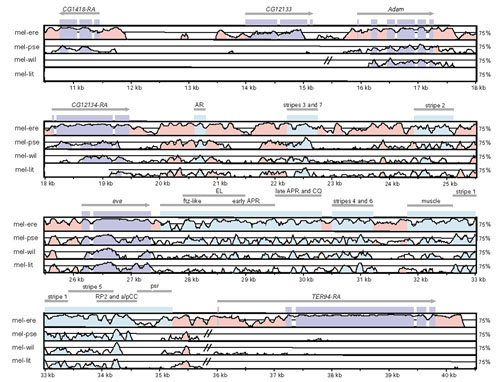
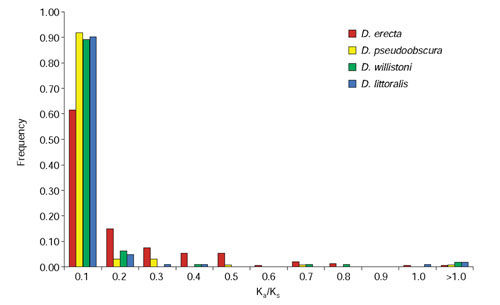
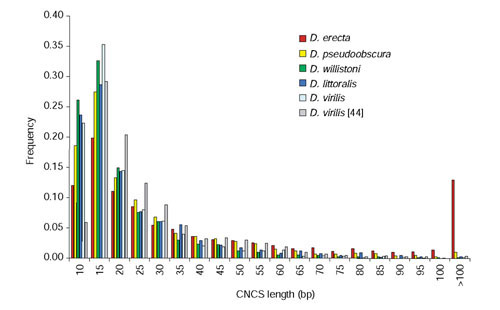
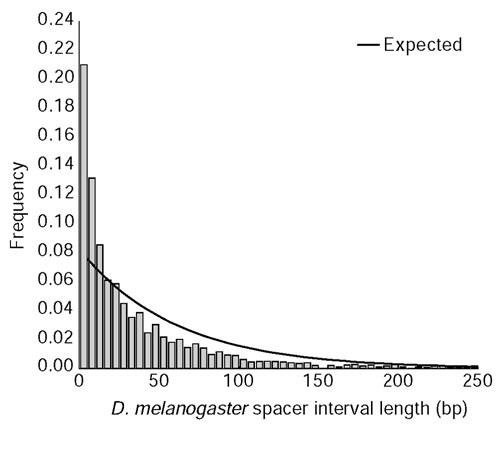
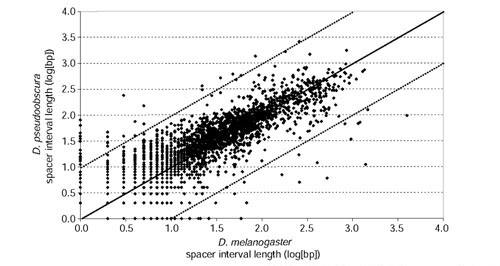
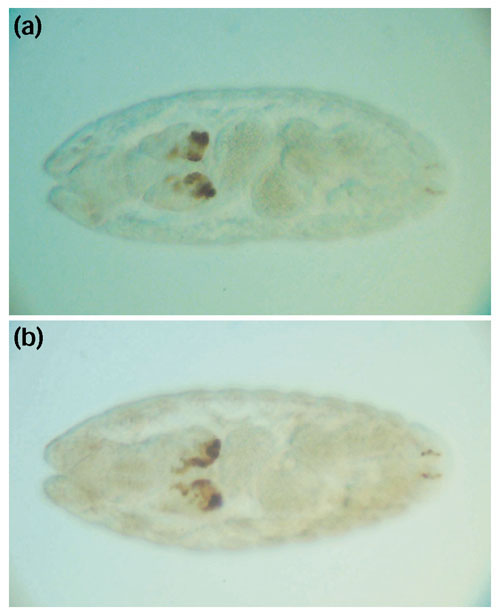
Results: We analyzed conservation in eight genomic regions (apterous, even-skipped, fushi tarazu, twist, and Rhodopsins 1, 2, 3 and 4) from four Drosophila species (D. erecta, D. pseudoobscura, D. willistoni, and D. littoralis) covering more than 500 kb of the D. melanogaster genome. All D. melanogaster genes (and 78-82% of coding exons) identified in divergent species such as D. pseudoobscura show evidence of functional constraint. Addition of a third species can reveal functional constraint in otherwise non-significant pairwise exon comparisons. Microsynteny is largely conserved, with rearrangement breakpoints, novel transposable element insertions, and gene transpositions occurring in similar numbers. Rates of amino-acid substitution are higher in uncharacterized genes relative to genes that have previously been studied. Conserved non-coding sequences (CNCSs) tend to be spatially clustered with conserved spacing between CNCSs, and clusters of CNCSs can be used to predict enhancer sequences.
Conclusions: Our results provide the basis for choosing species whose genome sequences would be most useful in aiding the functional annotation of coding and cis-regulatory sequences in Drosophila. Furthermore, this work shows how decoding the spatial organization of conserved sequences, such as the clustering of CNCSs, can complement efforts to annotate eukaryotic genomes on the basis of sequence conservation alone.
Figures

References
-
- Lewontin RC, Moore JA, Provine WB, Wallace B, Eds . Dobzhansky's Genetics of Natural Populations I-XLIII. New York: Columbia University Press; 1981.
-
- Patterson JT, Stone WS. Evolution in the Genus Drosophila. New York: Macmillan; 1952.
-
- Kreitman M. Nucleotide polymorphism at the alcohol dehydrogenase locus of Drosophila melanogaster. Nature. 1983;304:412–417. - PubMed
-
- Blackman RK, Meselson M. Interspecific nucleotide sequence comparisons used to identify regulatory and structural features of the Drosophila hsp82 gene. J Mol Biol. 1986;188:499–515. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
